- 首页 > 长读长人全基因组重测序
长读长人全基因组重测序
人类基因组变异的完整图谱对于深入了解遗传特征、助力疾病精准研究非常重要。虽然短读长测序可准确地检测SNP、InDel较小的变异类型,但对CNV和SV的检测灵敏度有限。近年来,长读长测序已被广泛用于人类基因组研究,其能够有效解析复杂的基因组结构,包括高度重复的基因区域和基因组结构变异,为寻找疾病相关变异提供了更全面的基因组视角。长读长技术的高准确性也使得它能够发现短读长测序技术可能错过的罕见变异,为精准医学基础研究提供更精确的基因变异信息。
产品亮点
- 类型全:全面覆盖各种变异类型
- 精度高:基于高精度测序数据,获得准确度和灵敏度更高的变异检测结果
- 成本低:基于PacBio Revio,以更低的成本获得更全面的数据
- 选择多:除标准SNP、InDel、SV、CNV外,还可提供复杂结构变异检测、甲基化信息
服务优势
- 读长长:超长的测序读长,可跨越高重复的区域
- 无偏向:单分子测序,无PCR扩增偏向性,避免了覆盖度不均一和系统误差
- 均匀覆盖:没有GC偏好性,可以轻松跨过高GC含量区域,保证序列的均匀覆盖度
- 变异检测更准确:变异检测灵敏度和准确度更高
- 通量高可量产:通量高,可量产,效果好
- 大项目经验丰富:大人群、大队列项目经验丰富
- 一站式全流程服务:从样本到结果的全流程服务
- 专业团队、实力雄厚:PhD级别专业团队支持
产品应用
- 复杂结构变异
- 疾病研究
- 癌症研究
- 人群队列研究
技术路线
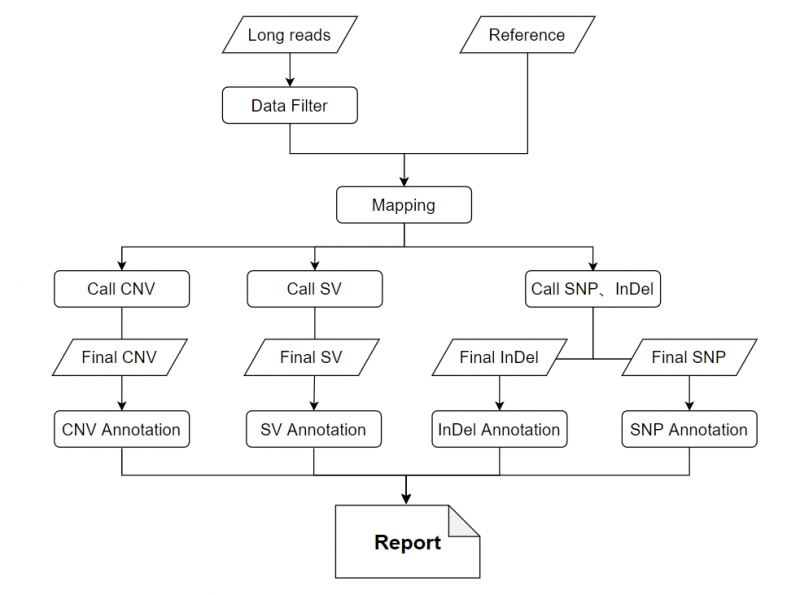
长读长测序的下机数据,获得到高质量的reads,将reads比对到参考基因组上,对比对结果进行排序得到的文件用于后续全变异检测,最后对变异结果进行注释及统计。
1、遗传病研究
案例一:Long-read sequencing to unravel complex structural variants of CEP78 leading to cone-rod dystrophy and hearing loss
2021年3月,Frontiers in Cell and Developmental Biology 期刊发表如题文章,研究结果表明,跨越CEP78 1-5的外显子缺失,在纯合与杂合状态下都会导致CEDHL功能丧失。本研究支持CRDHL具有极高的遗传异质性,并强调了SV分析在CRDHL遗传过程中的重要性。
之前的研究结果表明,CEP78基因结构变异可能会导致CRDHL(cone-rod dystrophy with hearing loss,视锥杆细胞营养不良与听力损失)。本研究利用长读长技术,对四个无亲缘关系的CRDHL家系进行了测序,将测序数据与参考基因组hg38进行比对,获得了高质量SV。且支持CEP78位点易于微同源性、复制性SV形成,(复杂)SV分析应纳入CRDHL或非典型Usher综合征的分子遗传检测。
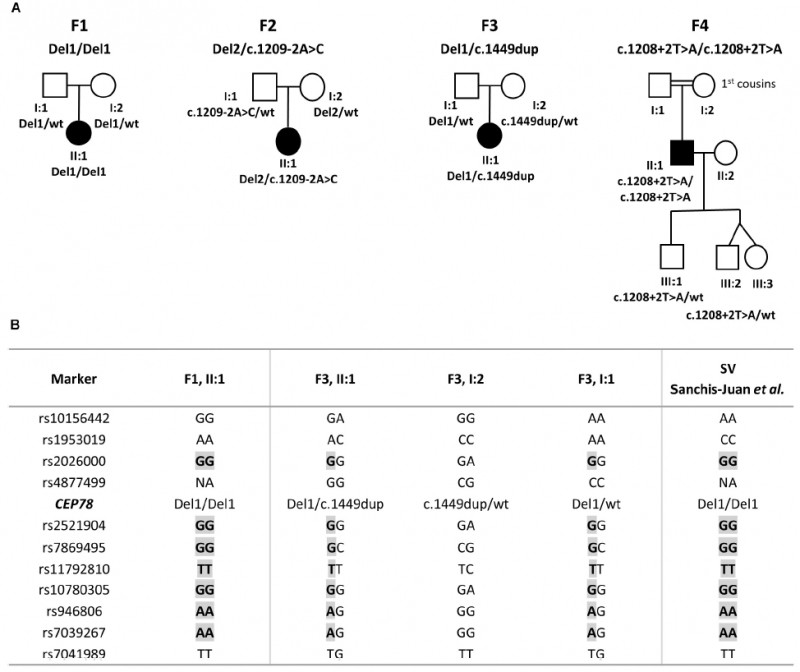
图1 分离CEP78结构和序列变异的家系和SV的单倍型重建
案例二:Familial thrombocytopenia due to a complex structural variant resulting in a WAC-ANKRD26 fusion transcript
2021年4月,Journal of Experimental Medicine发表了此文章。研究结果说明了传统基因组测序方法可能遗漏的复杂结构变异如何导致人类疾病。基因组测序技术的进步,已经发现了许多罕见疾病的病因。然而,许多案例仍未通过标准分子分析得到解决。文章描述了一个具有类似于遗传性血小板减少症2(THC2)表型的家庭。THC2通常由在造血分化过程中阻止ANKRD26表达沉默的单核苷酸变体引起。短读长全外显子组和基因组测序方法无法识别该家族中的因果变异。使用长读长全基因组测序,确定了一个涉及配对重复倒位的大型复杂结构变体。通过功能研究,表明这种结构变异导致致病性功能获得性WAC - ANKRD26融合转录本。
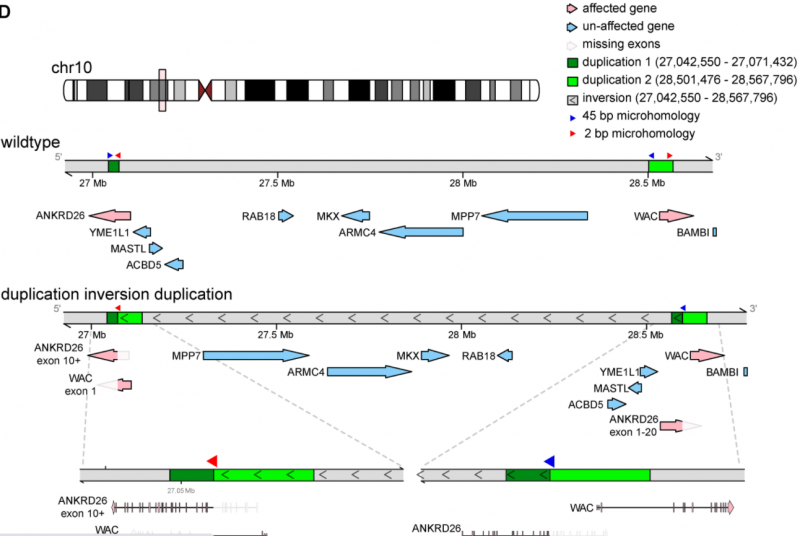
图2 野生型位点与成对重复倒置的局部组装
2、病毒插入研究
案例:The integration model of hepatitis B virus genome in hepatocellular carcinoma cells based on high-throughput long-read sequencing
2021年11月,Genomics发表了有关病毒插入研究的文章,本研究确定了HBV的整合模式,为通过基因编辑技术有效删除HBV序列提供了依据,也为病毒整合机制研究提供了新的导向策略。
肝细胞癌(Hepatocellular Carcinoma,HCC)是目前常见的恶性肿瘤之一,之前之前的研究发现,近半HCC病例与乙肝病毒(Hepatitis B virus,HBV)整合相关。本文首次采用长读长测序技术,对肝癌中病毒整合的完整模式进行了研究,直观地解析出病毒整合插入的完整序列,以及两端端点与结构变异的密切关系。
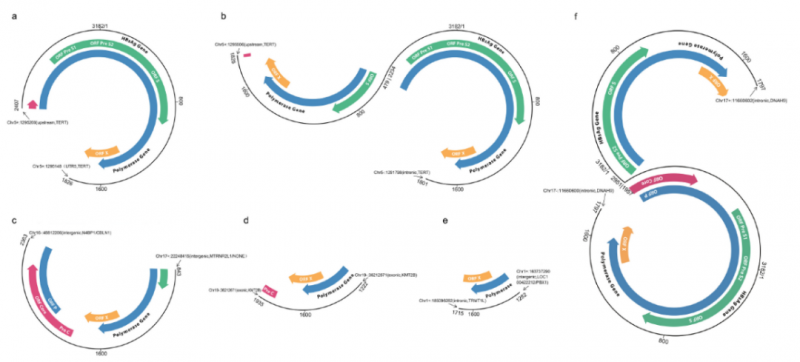
图3 HBV病毒热点研究区域整合模式研究
3、肿瘤疾病研究
案例一:Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer
2021年4月,Genome Medicine发表了此文章。本研究对之前研究过得DNA样本进行了重新测序,证明了长读长测序技术在分析人类多态性和体细胞突变方面的优势,揭示了癌症基因组复杂结构与变异的来源,为未来的遗传学研究做出了贡献。该研究利用长读长测序技术,对ICGG(International Cancer Genome Consortium,国际癌症基因组联合会)已报道的11例日本肝癌患者进行了全基因组测序,并与ICGC测序的正常样本进行了匹配,将测序数据与参考基因组GRCH38进行比对,获得高质量SV。由于测序reads较长,大多数reads被唯一map到参考基因组,本研究中长读长检测到的SV数量是短读长的1.6倍,表明长读长测序在检测SV方面更有效。对鉴定到的肝癌患者的体细胞SV进行分析,发现长读长测序技术可以检测到大量的体细胞SV与病毒整合,与鉴定到的生殖细胞SV比较后发现,体细胞与生殖细胞中的SV存在差异。
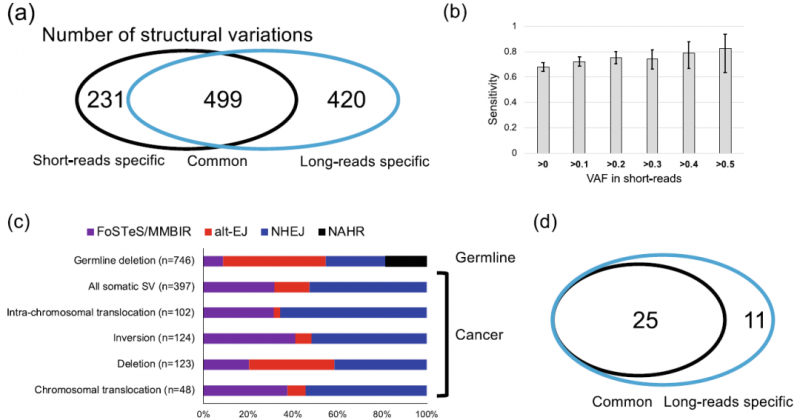
图4 肝癌中的体细胞结构变异
15X测序深度的PacBio Revio HiFi数据即可获得更优的全变异结果
长读长测序因其读长长,覆盖均一,且HiFi数据的精度高,能够大幅提升SV的可靠性和分辨率,并且SNP、Indel的检测灵敏度和准确度在15X覆盖深度的时候也已经超过短读长WGS 30X覆盖深度的水平。因此推荐至少15X以上的测序深度,深度越高灵敏度也会越高。
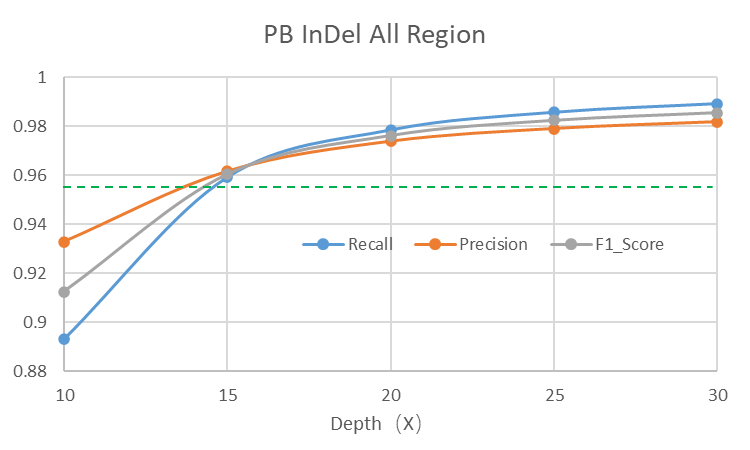
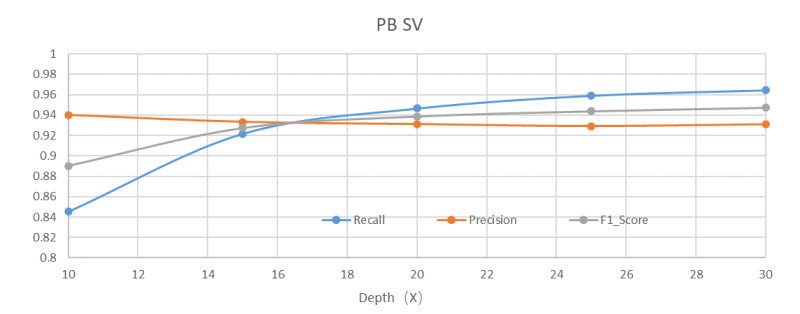
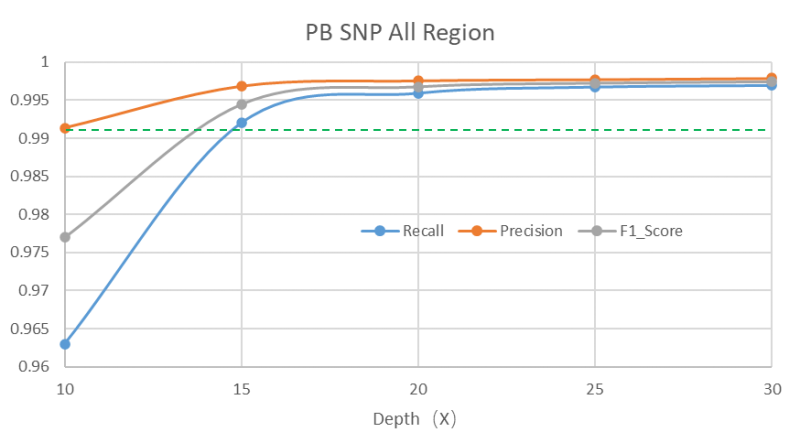
*上述分析结果由华大信息分析流程所得,测试结果不代表交付指标,最终解释权归深圳华大基因股份有限公司所有
- DNA送样建议

- 组织送样建议
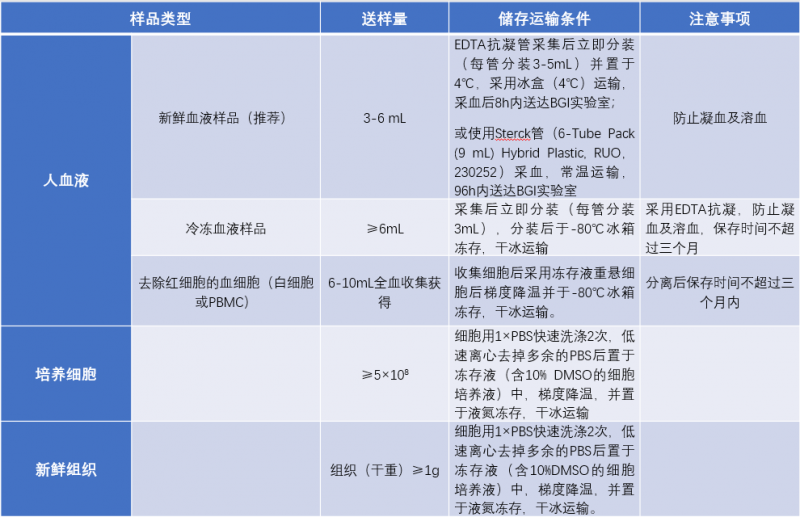
Q1: 如果自己提取,长片段DNA应该如何保存?
A1:对于提取后的长片段gDNA样本,4°C可保存半年,-20°C可保存一年,但-20°C保存建议只进行一次冻融,反复冻融会造成样本降解,样本 DNA 不能进行震荡或剧烈混匀操作,以免长片段DNA断裂。我们不推荐您送DNA样品,因为DNA的长度直接影响了phasing的长度。另外,DNA样品在运输途中会有损伤。
Q2:为什么要用长读长检测SV?
A2:结构变体(SV),包括缺失,插入,重复和倒位,占个体人类基因组中大多数变异的碱基对。许多研究中已经牵涉到人类健康的SV与相关表型有着直接或着间接的关系。因此,鉴定遗传变异并推断其功能影响是人类遗传学研究中最重要的问题之一。然而,因为SV易于在重复区域中出现并且内部SV结构可能出现的复杂性导致发现这些变体和基因分型变得具有挑战性。因此,需要长读长测序技术对SV结构进行新的探索,并对人类种群SV的检测进行分析,来理解与SV相关的疾病研究。
Q3:全变异和只做SV检测分别如何推荐策略?
A3:如果只做SV检测,ONT和PacBio都可以,测序深度推荐至少15X以上;如果是全变异检测,推荐Pacbio Revio平台的HiFi数据,测序深度推荐至少15X以上。