- 首页 > 宏基因组测序
宏基因组测序
产品介绍
微生物是地球上已知种类最多、数量最大、分布最广的生物类群,仅原核微生物的总量大约就达4×1030-6×1030个。但是传统的分离培养方法限制了认识微生物世界的视野。据估计,自然环境中超过99%的微生物不能用传统的方法进行纯培养,因而也不能对它们开展依赖于纯培养的生物技术或基础方面的研究。为了克服传统纯培养技术的不足,充分挖掘此类未培养微生物所蕴涵的巨大潜能,研究者们发展了宏基因组学的研究方法。利用分子生物学的研究方法绕过纯培养技术来研究微生物的多样性及功能,提供了一种探知微生物多样性结构和功能基因组的免培养方法,是一条寻找新基因及其产物的新途径。
研究内容
宏基因组研究以环境中所有微生物基因组为研究对象,通过对环境样品中的全基因组DNA进行高通量测序,获得单个样品的饱和数据量,基于denovo组装进行微生物群落结构多样性,微生物群体基因组成及功能,特定环境相关的代谢通路等分析,从而进一步发掘和研究具有应用价值的基因及环境中微生物群落内部、微生物与环境间的相互关系。构建的环境微生物基因集,可为环境中微生物的研究、开发和利用提供基因资源库。
产品优势
性价比高:自主测序平台,成本可控;
测序准确性高:DNBSEQ平台滚环扩增构建DNB测序文库,PCR扩增错误累积较少,高保真序列信息;
Duplication率低:DNBSEQ平台Duplication率低,同样的数据量有效数据多出3%-17%;
无index hopping担忧:DNBSEQ平台无index hopping担忧,结果更可靠;
经验丰富:有丰富的宏基因组项目经验,特别是在人体微生物研究方面处于领先地位,已发表文章100+,其中CNS系列文章26篇。
样本需求量低:常规宏基因组建库建议样本量在500ng以上;对于样本获取困难的样本,也可以选择微量建库,样本量可低至几ng。
合作模式:有专门的meta大项目团队,已发表多篇高水平文章。提供切实可行的项目方案,兼顾商业合作、科研合作优势。
技术流程
质量合格的基因组 DNA 样品通过超声波高性能样品处理系统(Covaris)随机打断,经过片段选择后得到 300bp左右的片段。加上接头,进行cluster制备,最后利用Paired-End的方法对插入片段进行测序,得到的原始数据经过质控和数据过滤,宏基因组组装、基因预测、构建参考基因集,并进行后续的物种、基因、功能分析。
建库流程
1) 将检测合格的基因组DNA样品用物理方法随机打断成300bp-400bp的片段;
2) 对打断的DNA片段进行末端修复,在3’端连接A碱基;
3) DNA单链环化;
4) 去除未环化序列并进行纯化;
5) 对构建的文库进行质量检测;
6) Make DNB;
7) 将质量检测合格的文库上机测序。
信息分析内容
|
分析模块 |
分析条款 |
|
1.
项目概览 |
项目信息 |
|
2.
数据过滤 |
数据过滤统计 |
|
3.
组装 |
Denovo组装 |
|
4.
基因预测及基因集构建 |
基因预测及基因集构建 |
|
5.
基因分析 |
1)
基因Venn图(2-5个样本或组别) |
|
2)
基因差异分析(分组≥2,每组样本数≥3) |
|
|
6.
基因集功能注释 |
Eggnog、KEGG、CARD、COG、CAZy、Swiss-prot,NR等 |
|
7.
物种分析 |
1)
物种累积曲线图分析 |
|
2)
物种组成分析 |
|
|
3)
物种差异分析(分组≥2,每组样本数≥3) |
|
|
4)
差异物种丰度热图(分组≥2,每组样本数≥3) |
|
|
8.
功能分析 |
1)
差异KO分析(分组≥2,每组样本数≥3) |
|
2)
差异KO丰度热图分析(分组≥2,每组样本数≥3) |
|
|
3)
差异pathway分析(分组≥2,每组样本数≥8) |
|
|
9.
多样性分析 |
1)
物种水平Alpha多样性分析 |
|
2)
KO水平Alpha多样性分析 |
|
|
3)
物种水平Beta多样性分析 |
|
|
4)
KO水平Beta多样性分析 |
|
|
10. 网络互作 |
1)
基于属水平的network分析 |
|
11. 相似性分析 |
1)
PCA分析 |
|
2)
PCoA分析 |
|
|
3)
NMDS分析 |
|
|
4)
基于距离热图分析(物种、KO) |
|
|
12. 关联分析与模型预测 (个性化) |
1)
CCA分析(需提供环境因子数据) |
|
2)
PERMANOVA/Adonis置换多元方差分析(默认分组因素对样品差异的影响,如关注表型对样品差异的影响需提供表型信息) |
|
|
3)
物种与表型Spearman相关系数分析(需提供表型信息) |
|
|
4)
MaAsLin分析(需提供表型信息) |
|
|
5)
随机森林--ROC曲线(分组=2,每组样本数≥30) |
|
|
13. 定制化分析 |
可结合客户的需求,协商确定信息分析内容 |
肠道微生物代谢的长链饱和脂肪酸可增强大鼠的结肠运动
Zhao L, Huang Y, Lu L, et al. Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats(Microbiome, 2018).
胃肠功能紊乱是患有功能性胃肠病和其他肠道疾病的患者中常见的一种主要症状,慢性或反复发作的胃肠运动障碍严重影响患者的生活质量。目前对胃肠功能紊乱的发病机制研究并不完整,这限制了胃肠功能紊乱的个体化和精准有效药物的开发。前期研究表明肠道菌群在胃肠运动紊乱中起关键作用,但肠道菌群与宿主胃肠道运动相互作用及其作用机制尚不明确。
新生儿母亲分离(NMS)的啮齿动物模型是以一个成熟的长期结肠功能障碍模型,主要表现是结肠动力增强;有研究表明小鼠和大鼠在受NMS处理后会出现肠道微生物紊乱;而NMS模型中肠道菌群变化与胃肠动力增强的关系仍有待研究。本研究通过大鼠NMS模型和无菌模型,研究肠道菌群在胃肠功能紊乱中的作用,并深入挖掘其作用机制。
研究方案:
1. NMS模型大鼠肠道代谢水平的改变
分组:对照组VS NMS组
研究方法:代谢组学
通过代谢组学检测NMS大鼠肠道代谢水平的变化,得到与NMS模型大鼠胃肠动力改变相关的代谢产物(SLCFA)。
2. 实验验证代谢水平的改变对大鼠胃肠的影响
体外实验:通过器官浴系统研究不同剂量C17:0,C18:0以及游离LCFA受体拮抗剂对体外结肠环状肌收缩的影响;
体内实验:不同剂量C17:0和C18:0对正常大鼠(n=8/group)胃肠动力的影响
3. NMS模型大鼠肠道菌群及功能的改变
分组:对照组VS NMS组
研究方法:宏基因组学
通过宏基因组学检测NMS大鼠肠道菌群及基因变化,得到与对应代谢产物(SLCFA)相关的菌群及基因
4. 验证肠道菌群改变对胃肠运动及代谢产物的影响
伪无菌大鼠模型:将对照组和NMS组大鼠粪便分别移植到伪无菌大鼠体内,并检测移植后大鼠的粪便菌群、基因及胃肠运动和对应代谢物水平的改变
抗生素治疗:新霉素调节NMS大鼠肠道菌群,以及治疗后胃肠动力、肠道菌群、代谢水平的变化
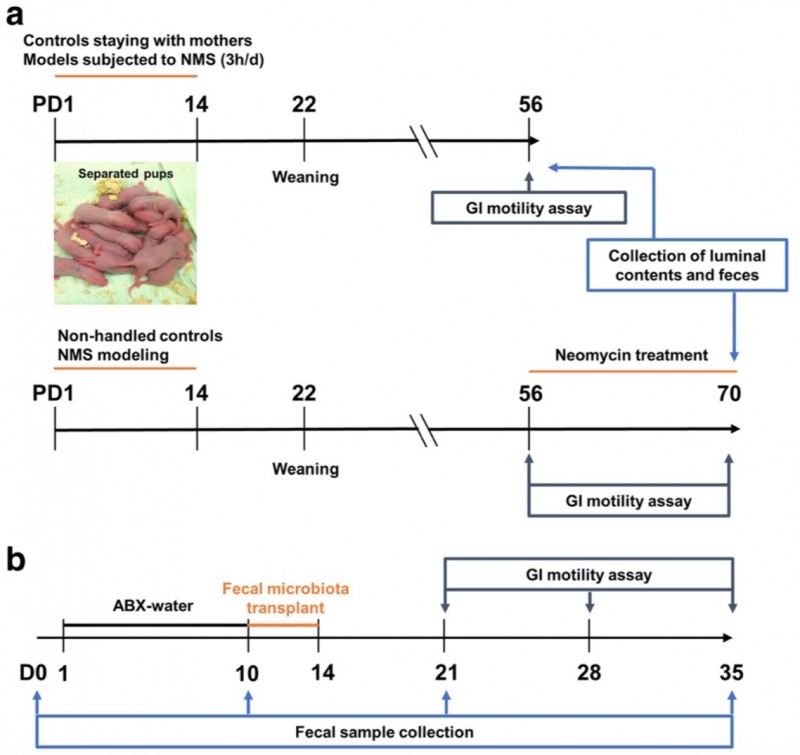
图1. 实验方案。a, 两窝大鼠,对照组VS NMS组,胃肠动力检测,肠道和粪便菌群检测及粪便代谢物检测;NMS 新霉素处理组和对照组;b, 无菌大鼠粪菌移植及肠道菌群检测。
主要结果:
1. 代谢组分析发现结肠中过量的SLCFAs与NMS组肠动力增强有关。体内体外验证结果表明SLCFAs刺激大鼠结肠收缩并增强排便频率
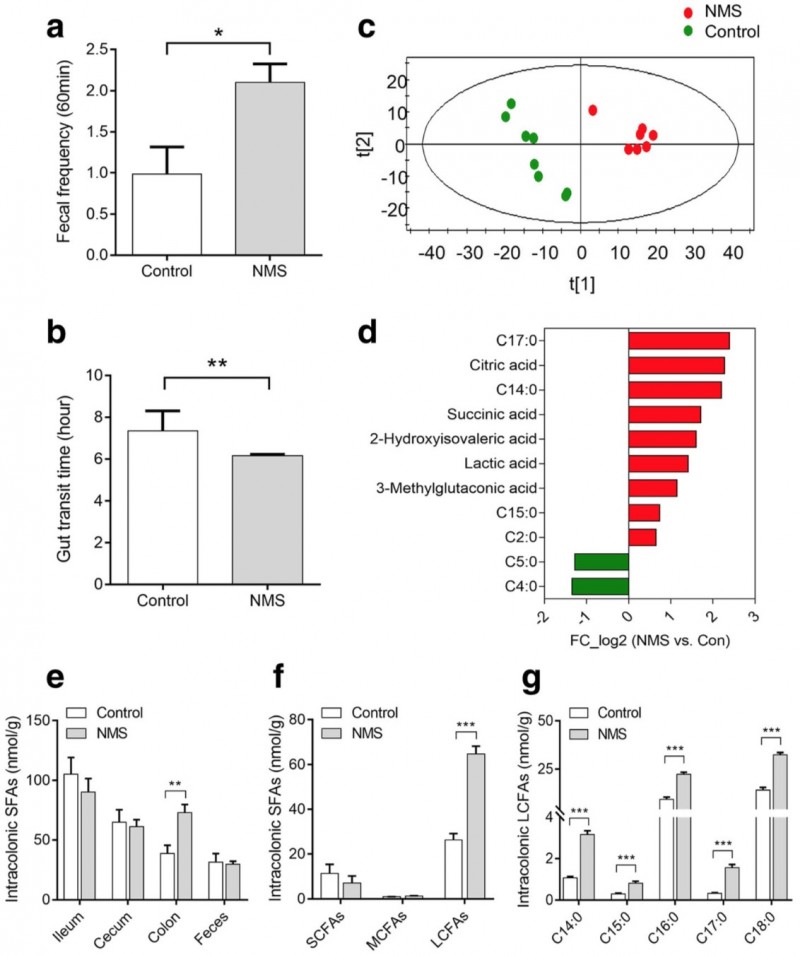
图2. 与对照组相比,NMS大鼠的肠道运动性增强,结肠内饱和长链脂肪酸(SLCFAs)含量增加。
2. 宏基因组分析发现NMS组中差异的粪便微生物与SLCFAs生成相关。粪便移植验证结果表明NMS供体的粪便微生物使无菌小鼠排便频率及结肠内SLCFAs含量增高。用新霉素改善NMS组肠道菌群失调可有效降低肠动力及SLCFAs产生。
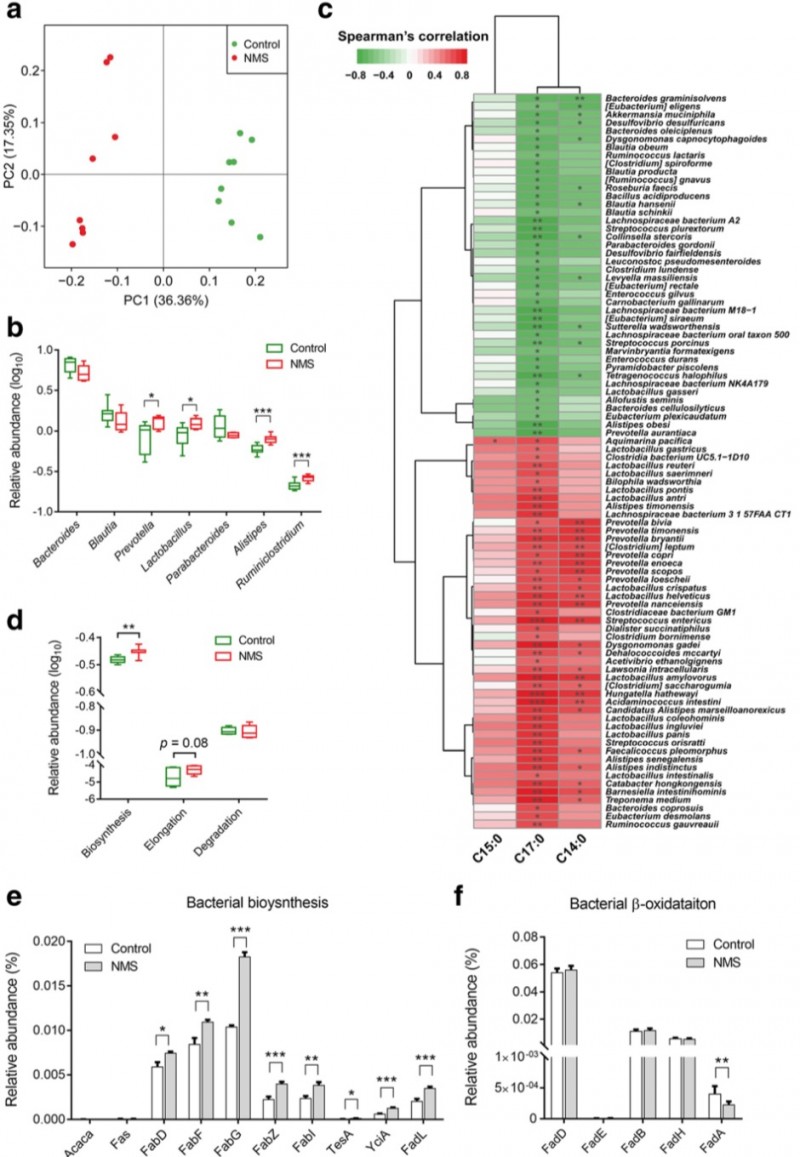
图3. 粪便微生物组的改变与NMS大鼠中的SLCFAs生成密切相关。
降糖药疗效和肠道菌群特征关系的研究
Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment(Nature communications, 2017).
众所周知,小肠α葡萄糖苷酶抑制剂阿卡波糖已在糖尿病患者,尤其是中国患者的治疗中得到了广泛应用。相比传统的磺脲类药物,阿卡波糖可以为糖尿病患者带来更多代谢获益,如体重下降、血脂谱改变等。除了降糖以外,是否还有其他机制可以解释抗糖尿病药物的代谢获益?不同患者对于不同降糖药的反应为何各不相同?肠道共生菌群是否在其中发挥作用?
本研究通过多中心、随机开放的临床研究通结合肠道宏基因组及血浆、粪便代谢组学发现:常用降糖药物阿卡波糖可显著改善初发II型糖尿病患者肠道共生菌以及肠道共生菌胆汁酸代谢水平;并且肠道内富含拟杆菌的患者接受阿卡波糖治疗后会表现出更多代谢获益,如减重、降脂以及改善胰岛素抵抗。本研究首次建立了降糖药疗效和肠道共生菌群特征关系的研究,这一发现不仅破解了阿卡波糖降糖外的代谢获益的机制之谜,同时也为设计靶向肠道共生菌胆汁酸代谢的新型糖尿病药物提供了新的研究思路。
样本来源:
分组:阿卡波糖组(n=51),格列吡嗪组(n=43);
样本类型:粪便样本(meta、代谢组)、血浆(代谢组)
研究方法:
宏基因组、代谢组学
主要结果:
1. 阿卡波糖和格列吡嗪两种均有较好的糖尿病治疗效果。在等效降糖情况下,阿卡波糖相较格列吡嗪具备更多改善患者体重、胰岛素抵抗以及血脂的疗效。
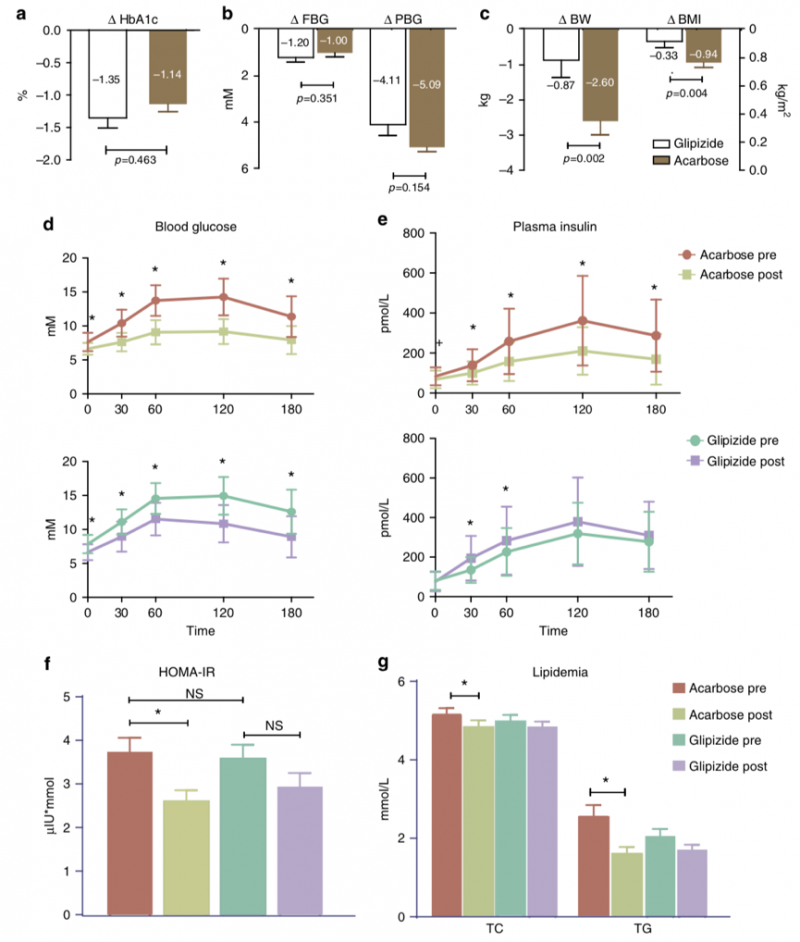
图1 糖尿病患者阿卡波糖或格列吡嗪治疗前后,相关临床指标的变化
2. 阿卡波糖治疗组胆汁酸代谢变化发生明显的改变。这些代谢指标的变化与糖尿病临床指标相关。

图2 阿卡波糖治疗前后胆汁酸代谢的变化
3. 与磺脲类药物相比,阿卡波糖可以显著调节患者的肠道菌群的结构与功能。阿卡波糖治疗后,现场到菌群的基因和吴总数目均有减少。在物种层面,阿卡波糖治疗可显著提高多种益生菌(如双歧杆菌和乳酸菌)丰度,并大幅降低梭菌和拟杆菌丰度。在功能层面,一方面,双歧杆菌和乳酸菌替代拟杆菌成为胆盐水解酶(BSH)基因的主要提供者;另一方面,合成疏水性次级胆汁酸的关键限速酶(7-α/β类固醇脱氢酶)的编码基因丰度在治疗后大幅度降低。这些肠道菌群的改变提示,由肠道微生物合成的肝毒性的疏水性次级胆汁酸(脱氧胆酸和石胆酸)生成减少;有益的亲水性次级胆汁酸如熊脱氧胆汁酸生成得到促进。

图3 阿卡波糖治疗显著影响肠道菌群的基因和物种结构。a-d,基因数目变化;e, 物种变化。

图4 阿卡波糖治疗影响次级胆汁酸代谢
a,阿卡波糖治疗后,次级胆汁酸代谢酶发生显著变化;b, 阿卡波糖治疗前后提供bsh基因的菌群结构发生了变化;c, 阿卡波糖治疗对胆汁酸代谢通路的影响。
DNA样本要求
|
Metagenomic Survey |
||||
|
样本类型 |
总量 |
浓度 |
完整性(胶图) |
纯度 |
|
Genomic DNA |
1μg |
12.5ng/μl |
主峰>20Kb |
无蛋白,RNA/盐离子等污染,样本无色透明不粘稠 |
组织样本要求
|
组织类型 |
宏基因组测序 |
|
Meta (粪便) |
≥200mg |
|
Meta (土壤) |
≥1000mg |
对于Meta类样品提取效果,取决于样品中的微生物丰度,丰度低的样品类型,极有可能无法提取到核酸。
常见样本取样方法
液氮速冻法
① 人和大型动物粪便样品的采集
a) 准备好便盆和粪便容器,洗手,带上手套收集新鲜的粪便样本;
b) 在实验室将装好受试者粪便样本,立即进行分装并标记;
c) 用无菌牙签或粪便取样器截取样品中段里部(粪便表层含有肠粘膜脱落细胞;
外部容易污染,且接触空气后,部分细菌 DNA 开始降解),取约50~100mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取3-5管备份;
d) 分装好后,立即液氮速冻或直接放入-80℃低温保存,送样时选择干冰运输寄送。
注意:如粪便样品量较多或不能马上冻存,最迟要在2小时之内全部收集完。
② 小型动物(如鼠)颗粒粪便样品采集
a) 动物颗粒粪便样品,动物排便后立即装入2.0mL离心管中(小鼠粪便3粒每管),放入-80℃低温保存,送样时选择干冰运输寄送。
③ 肠道内容物样品采集
a) 用无菌解剖刀,在无菌状态下取出整个肠道,切取所需肠段的内容物(条件允许的话,可在无菌操作台进行);
b) 用无菌手术刀挖取内容物,装入无菌2.0mL离心管中,每管取约50~100mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取3-5管备份;
c) 分装好后,立即放入 -80℃低温保存,送样时选择干冰运输寄送。
④ 肠道组织样本采集
a) 组织样本用无菌磷酸盐缓冲液轻轻清洗,直到没有内容物流出;
b) 用无菌的显微镜玻片刮取附着在表面的组织细菌,转移到无菌的2.0离心管中;
c) 立即转入-80℃低温保存,送样时选择干冰运输寄送。
⑤ 土壤meta样品采集
a) 根据研究目的确定采样范围,取样器具要事先消毒灭菌处理,开始采样;
b) 去除表面浮土,使用乙醇火烧的铲子挖取地下5~20cm的土层;
d) 去除可见杂质后,土壤过2mm筛网,建议每个样品从3个及以上采样点采集并混合而成,把土样装入无菌2.0mL离心管中,每管取约200mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取5管以上备份;
c) 分装好后,立即转入-80℃低温保存,送样时选择干冰运输寄送。
⑥ 水体样本采集
a) 根据研究目的确定采样深度和范围;
b) 采集好的水样需要通过滤膜进行过滤,可以根据水样的浑浊程度选择相应孔径的滤膜;
c) 将滤膜转移到2.0mL离心管中,立即转移至-80℃低温保存,送样时选择干冰运输寄送。
清亮水样:可选择小孔径的滤膜,一般选0.22μm或0.45μm的滤膜,过滤水样体积大于10L;
浑浊水样:过滤前静置分离悬浮颗粒,也可以用大孔径的滤膜预过滤一遍,再用小孔径的滤膜进行过滤。
商业核酸保护液保存法
人粪便样品可采用常温采样套装,具体请按照说明书操作保存运输样品。
Q1. 如果存在较大的宿主的污染,且没有宿主基因组的参考序列可以进行宏基因组测序吗?
不可以,如果宿主的基因组序列在环境DNA中的量比较多,测序之后,我们没有办法通过已知的宿主基因组的序列来去污染,会对最后的分析结果造成很大影响,而且可用的数据量会很少;但是如果在提取的过程,宿主基因组的污染的量很少,后期的数据分析还是可用的,但是会存在一定的风险。
Q2. 进行多样品比较分析的条件?
样品数量超过2个,且有比较意义的,均可进行多样品比较分析。但若要对明显分组的样品进行比较分析,建议至少2个组(每组至少10个样品以平衡个体差异)。
Q3. 不同环境样本数据量要求?
一般推荐简单环境(如哺乳动物肠道)测序数据量为5G clean data;复杂环境(如土壤、海洋等)推荐数据量为10G clean data。
Q4. 宏基因组和16S研究主要有哪些区别,这两种产品应该如何选择
宏基因组和16S研究的差异:
1)研究内容:
宏基因组测序是对环境中所有微生物进行全基因组测序,可以研究群落的物种、基因、功能结构/差异等;
16S测序是对细菌的16S保守区域进行测序(真菌测18S或ITS信息),可以根据16S/18S/ITS序列的差异性进行物种分析,或者进行群落物种鉴定。也可以进行物种功能预测,该功能预测在功能差异分析中往往结果较差。
2)覆盖范围:
宏基因组研究理论上可以检测环境中所有生物的基因组信息(包括原核、病毒、真核生物信息),环境中其他DNA污染会影响测序结果;
16S测序过程首先对目的区域进行扩增,只针对细菌(18S/ITS针对真菌)进行研究,测序过程受其他物种DNA影响较小。
3)样本量需求
宏基因组研究对样本量要求较高,一般要求500ng以上,低于500ng也可以进行微量建库,不建议同一个项目同时选择常规建库和微量建库。
16S测序只要能扩增出目的片段即可建库测序。
4)测序策略
宏基因组建库片段大小在300bp左右,不需要完全测通,目前多采用PE150;
16S测序策略依据目的片段大小而定,需要对目的片段完全测通,一般选择PE250或PE300测序;目的片段更长的需要选择长读长测序。
如果客户主要关注群落物种信息,或者是做菌种鉴定或希望发现新物种,推荐选择16S测序;如果客户希望挖掘群落相关基因或功能信息,则推荐选择宏基因组测序。此外在大样本研究中,为了节约成本,也可以先选择16S进行初筛,然后选择关键样本或组别进行宏基因组测序,进行基因或功能挖掘。
Q5. 宏基因组研究推荐多少样本量
宏基因组样本量需求跟研究目的直接相关。如果侧重于功能挖掘,一个或几个样本即可;如果侧重于组间差异分析,如疾病相关的肠道菌群研究,一般需要较大的样本量,一般每组样本在50个左右,可参考对应的经典研究案例。
Q6. 对于有参考基因集的微生物群落研究(如人肠道微生物、小鼠肠道、猪肠道),是否可以直接比对已有的参考基因集进行物种、基因注释,这种操作有什么优势/劣势?
可以。
优势:已经公开发表的参考基因集一般是基于大样本量数据构建而成,基因集覆盖范围广,质量高;直接比对已有的数据库不受单次测序数据质量的影响,结果准确,且信息分析周期较短。既适用于一般的商业项目,也适用于大型合作项目初筛。
劣势:公开发表的参考基因集虽然覆盖范围较广,来源特殊的样本,可能和参考基因集差别较大;在大型研究中,可能也会忽略掉群落特异的(新的)基因或物种产生的影响;因此在大型项目中,可以先基于已发表的参考基因集进行初筛,同时基于新的项目数据进一步完善已发表的参考基因集。