- 首页 > RNA-Seq
RNA-Seq
RNA-Seq,对某一物种或特定细胞在某一功能状态下产生的mRNA进行高通量测序,可以提供定量分析,检测基因表达水平差异。
技术优势
数字化信号:测序直接获得序列,无背景噪音,无交叉杂交;可鉴别序列间单个碱基的差异;
高通量:一次测序得到千万条以上的序列;
检测阈值宽:跨越6个数量级的宽检测阈值,从几个到数十万个拷贝精确计数;
良好的重复性:深度测序保证了抽样随机性,重复性非常好,无需重复实验;
高灵活性 :依据客户需求,提供不同测序平台服务。
产品应用
医学上的应用方向——
农学上的应用方向 ——
研究内容
标准信息分析:
1. 基本数据统计:① 去除接头序列、低质量序列得到reads信息,② 样品相关性,③ 表达量分布,④ RNA分类;
2. 参考基因组比对;
3. mRNA鉴定;
4. mRNA定量分析;
5. mRNA差异表达分析(样本间、组间);
6. mRNA表达/差异基因聚类;
7. mRNA差异基因GO分类、富集;
8. mRNA差异基因KEGG分类、富集。
Dr.Tom信息分析:
一)数据库注释
1. 转录因子注释(AnimalTFDB/PlantTFDB);
2. GSEA分析;
3. Rfam、Pfam、Reactome、COG、EggNOG和InterPro数据库注释;
二)互作网络分析
1. 靶基因分析① miRNA-mRNA靶向关系分析,② lncRNA-mRNA靶向关系分析;
2. ceRNA互作网络分析;
3. 蛋白互作网络分析;
4. 共表达互作网络分析;
三)特色分析
1. 自定义标签和自有数据上传;
2. 外部数据库信息(TCGA、ARCHS4);
3. 关键驱动基因网络图分析;
4. 时间序列分析。
(*以上分析内容为部分物种可做。)
定制化信息分析:
1. 可结合客户的需求,协商确定定制化信息分析内容。
医学案例:BGISEQ RNA-Seq揭示癌症CAR-T免疫细胞疗法中T细胞耗竭主因
案例描述:
CAR-T疗法是通过基因工程对患者自身的免疫T细胞进行改造,插入识别癌细胞抗原的嵌合抗原受体(CAR),使其具备攻击癌细胞的能力,然后输注回患者体内产生免疫应答,消灭癌症。
CAR-T细胞疗法近年来成为癌症免疫治疗最热门的领域之一,但T细胞耗竭使疗效不持久。为了解决这一新兴治疗方式的主要障碍,本文开展研究,揭示了耗竭的关键是转录因子c-Jun功能缺陷,通过对CAR-T细胞进行改造可以有效增强其抗衰竭能力。DNBSEQ平台提供RNA-Seq。
研究结果:
在本研究中,研究人员通过可调节性信号CAR模型,找到T细胞耗竭的显著特征。T细胞耗竭与IL-2产生过程出现缺陷有关,伴有AP-1转录因子motif染色质开放性增加以及bZIP和IRF转录因子过表达。从图5可以看出,跟对照相比,耗竭T细胞表现出不同的基因表达模式(e, f)。ATAC-Seq的结果表明,耗竭T细胞中鉴定得到染色质开放区域远高于对照(g, i, j)。从图6可以看出,AP-1转录因子的表观遗传和转录失调。
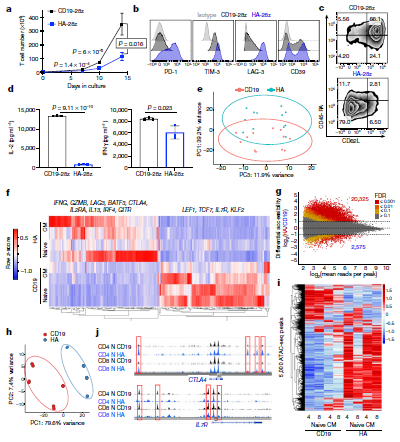
图1 HA-28z CAR T细胞表现出T细胞衰竭的表型、功能、转录和表观遗传特征
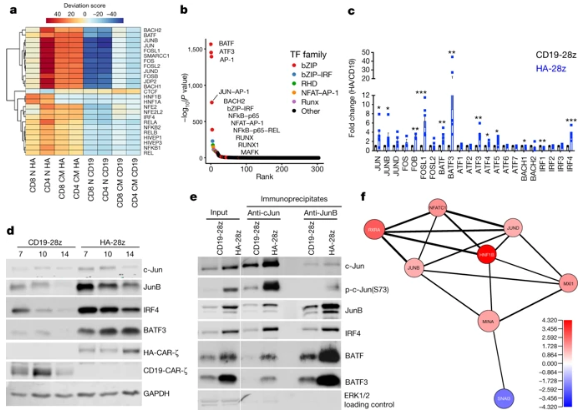
图2 耗竭的CAR-T细胞中的AP-1家族标志
参考文献
Lynn R C, Weber E W, Sotillo E, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance[J]. Nature, 2019: 1-8.
农学案例:BGISEQ RNA-Seq作物高效利用氮肥调控机制研究
案例描述:通过选育半矮化作物,极大地提高了绿色革命品种谷物的产量,但因使用过多无机氮肥而导致环境恶化日益加剧。为加强全球粮食安全及可持续发展,提高作物氮利用效率的需求日益迫切。因此,亟需深入了解作物生长,氮同化、碳固定的共同调节机制。
研究方法:以NJ6 (SD1)为对照,对36个含有sd1的籼稻品种分析NH4+的利用率。再以NJ6为轮回亲本与一个高NH4+吸收率品系NM73杂交创建BC1F2群体,通过QTL定位、图位克隆等技术获得氮肥高效利用的关键基因。分析关键基因启动子的三种SNP分型,筛选不同型别影响作物产量的潜在联系。
研究结果:
1、GRF4-DELLA相互平衡作用调节植物的生长和代谢。GRF4是生长调节因子,调节促进植物氮代谢和稳态,也协调促进植物碳代谢和生长,而DELLA生长阻遏物抑制这些过程。
2、半矮化作物中,在适度的氮素水平下增加GRF4的表达,GRF4-DELLA平衡机制偏向于GRF4的促进作用,从而利于氮、碳的吸收,增加叶、茎的宽度,但对高度影响很小。因此,GRF4表达增加改善氮素吸收效率的同时,不影响半矮化性状,能进一步提高作物产量。
结果展示:
通过基于BGISEQ平台的RNA-Seq和ChIP-seq两项技术,解析GRF4作用的分子机制,证明GRF4是激活下游参与氮素吸收和氮素同化的相关基因。ChIP-Seq揭示了潜在的GRF4靶识别位点,其中主要是多个氮代谢基因启动子共有的GGCGGC-motif。
GRF4与GIF1(GRF-interacting factor 1)复合体结合包含GCGG-motif的启动子,促进氮素同化吸收。SLR1(属于DELLA家族)在该途径中可抑制GRF4与GIF1的相互作用,导致氮素利用率降低。同时,GRF4还可以促进碳同化吸收相关基因的表达。
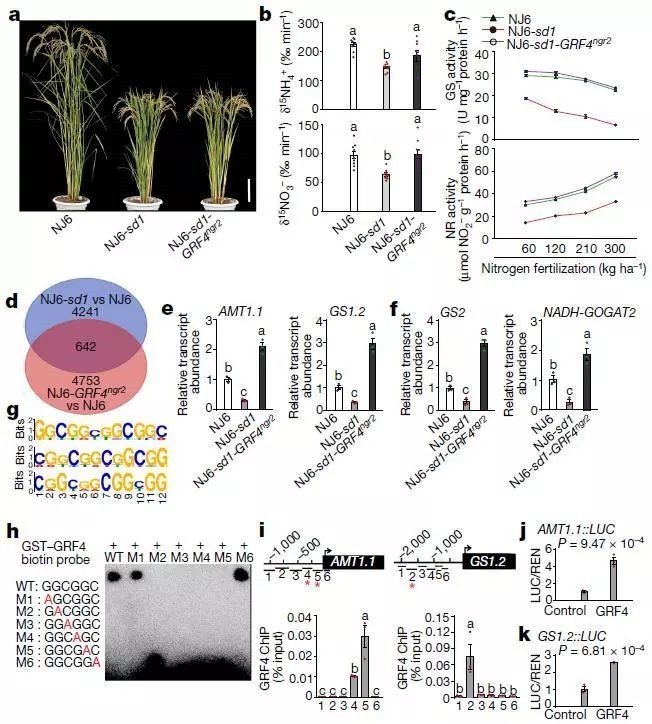
图3 GRF4调控多种氮代谢基因的表达
参考文献
Li S, Tian Y, Wu K, et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature. 2018 Aug;560(7720):595-600.
部分内容展示:
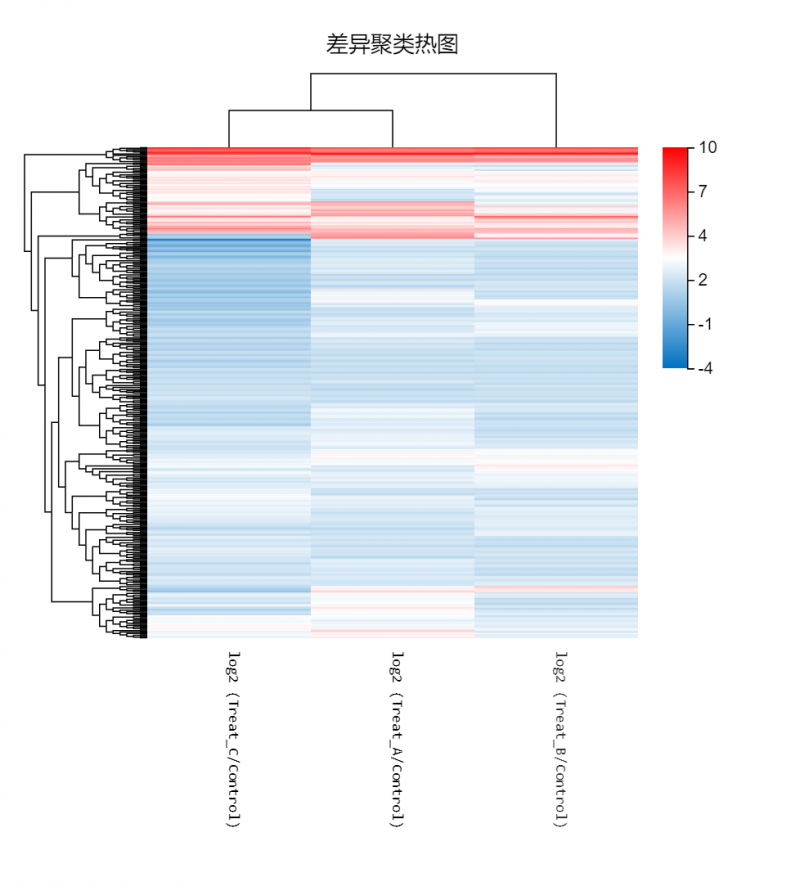
图1 差异聚类热图
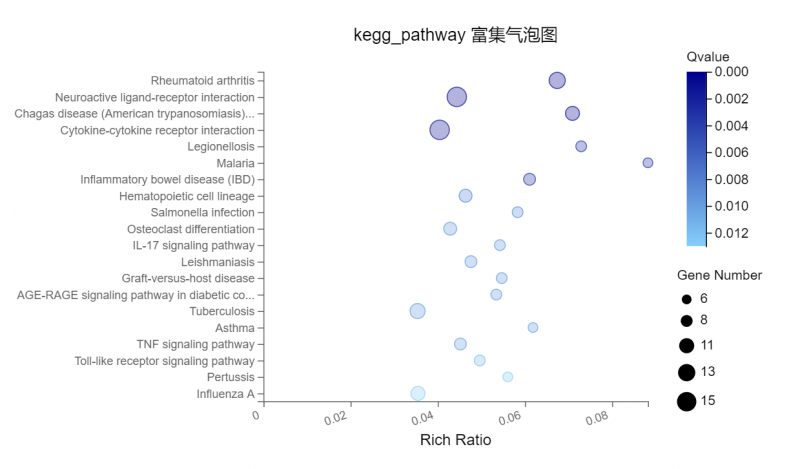
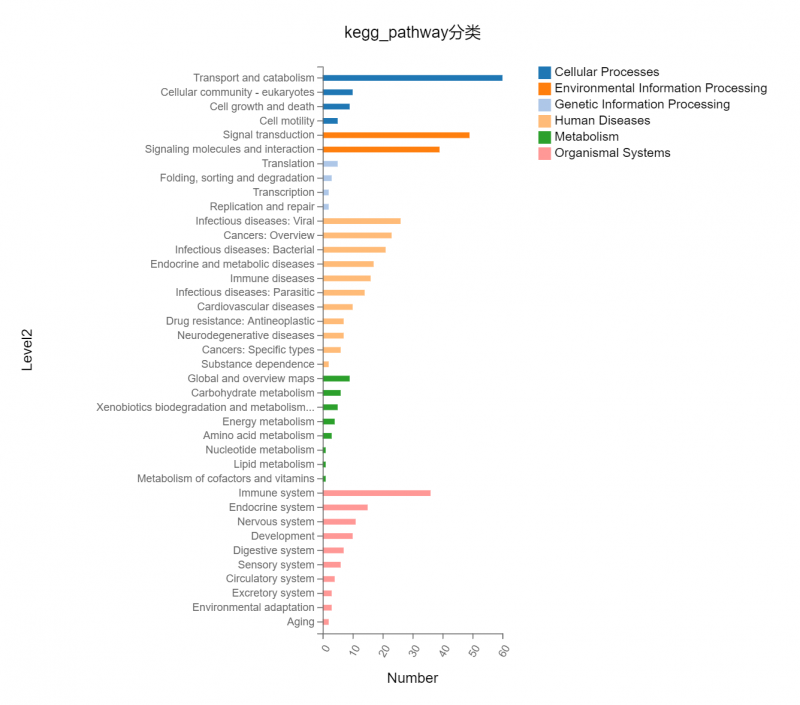
图2 功能富集或分类图

图3 蛋白互作网络
表1 RNA-Seq核酸样品判定标准
|
RNA-Seq (Quantification)(默认链特异性, 非链特异和链特异性送样建议相同) |
||||||
|
样本类型 |
总量 |
浓度 |
RIN |
28S/18S (23S/16S) or DV200 |
基线和 5S |
纯度 |
|
Total RNA(真菌) |
≥1μg |
20-1000 ng/μL |
RIN≥6.5 |
28S/18S≥1.0 |
基线平整, 5S 峰正常 |
无 DNA,蛋白/盐 离子等污染,样本 无色透明不粘稠
|
|
Total RNA (植物) |
≥400ng |
10-1000 ng/μL |
RIN≥6 |
28S/18S≥ 1.0 |
||
|
Total RNA(动物) |
≥400ng |
10-1000 ng/μL |
RIN≥7.0 |
28S/18S≥1.0 |
||
|
Total RNA (非全血人鼠)
|
≥200ng |
10-1000 ng/μL |
RIN≥7.0 |
28S/18S≥1.0 |
||
|
Total RNA(昆虫) |
≥400ng |
10-1000 ng/μL |
N/A |
|||
表2 RNA-Seq 组织样品判定标准
|
组织类型 |
RNA-Seq文库 |
|
新鲜培养细胞 (细胞数) |
≥2×105cell
|
|
新鲜动物组织干重 |
≥30mg
|
|
新鲜植物组织干重 |
≥100mg
|
|
全血 |
≥1
mL 全血收集的淋巴细胞或 ≥1 mL Paxgene Blood RNA tube / RNAprotect ® Animal Blood Tubes收集的全血 |
|
菌体(细胞数或干重) |
≥2×105cell
或 ≥30mg |
|
FFPE |
≥5片,未染色,100mm2,5~10μm 厚度 |
Q1:RNA-Seq是否需要做生物学重复?如果需要,一般要多少个重复样本?
答:是的,2011 年 7 月 Hansen[1]发表的文章表明生物学差异是基因自身表达的特性,与检测技术的选择以及数据处理的方式无关。生物学重复实验,可以在很大程度上消除样本的个体差异、系统平台差异,能更准确地检测差异基因。如果不设生物学重复,高影响因子的杂志可能会遭编辑质疑。不建议2个生物学重复是因为,如果两者结果不一致,无法确定以哪个数据为参考。3个生物学重复,如果出现1个结果不一致的,可以取另外2个的结果。
Q2:RNA-Seq必须要有参考基因组序列吗?
答:不是必须要有参考基因组序列,但一定要有参考序列,如unigene序列、mRNA序列、CDS序列等可以作为参考序列。也可通过参考有基因组序列的近缘物种的基因注释信息来完成相应的标准信息分析,不过此种方法并不推荐。
Q3:RNA-Seq如果没有参考序列怎么解决?
答:推荐做转录组de novo组装获得unigene作为RNA-Seq参考序列。需要注意的是,要把所有做RNA-Seq的样品,都混合成一个样品,做转录组。
Q4:RNA-Seq推荐测序数据量与基因组大小有关吗?
答:RNA-Seq推荐的测序数据量,主要与基因数量有关,不同物种基因组大小相差比较大,但是编码基因的数量相差并不大,一般物种在3万左右。所以对于一般物种的RNA-Seq数据量,10M clean reads是足够的。一般HiSeq平台推荐10M clean reads数据量,BGISEQ-500平台,为了给研究者更准确全面的数据结果,提供20M clean reads,而价格比HiSeq平台10M clean reads数据量还低。
Q5:有些观点认为RNA-Seq测序策略是SE100,有必要测这么长吗? PE测序是不是会比SE测序更好?
答:没有必要,SE50足够。做RNA-Seq,只要将得到的reads比对到参考基因集,得到基因ID就可以了,不需要那么长,因为不需要做组装,也不需要做定性分析。文献支持:康奈尔大学维尔医学院的研究人员在Genome Biology杂志上发表文章[2],认为对于差异表达分析而言,读长并非越长越好。研究人员认为,在RNA-seq研究中使用什么样的读长,这要取决于研究的最终目的。如果只需要差异表达基因,那么50 bp的单端读取就够了。与100 bp的双端读取相比,使用50 bp的单端读取能够节省成本、时间。
Q6:测序后有何验证方法?
答:实验验证的方法最常见的是通过实时荧光定量 PCR(qRT-PCR)技术来验证测序结果。还有FISH(原位荧光杂交)、微阵列芯片技术、Northern blot等。 功能验证一般是基因敲除、敲低或过表达,转基因等。
Q7:BGISEQ-500 RNA-Seq有没有发布demo数据?
答:已发布一个UHRR人标准品下机数据并共享了数据结果和分析方法,下载请点击这里>>
参考文献
1. Kasper D Hansen, Zhijin Wu, et al. Sequencing technology does not eliminate biological variability. Nature Biotechnology. 2011. 29(7): 572–573.
2. Chhangawala S, Rudy G, Mason CE, et al. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biology. 2015 Jun 23;16:131.