- 首页 > 全长转录组
全长转录组
产品介绍
基于短读长测序平台的转录组由于读长的限制(PE100/PE150),在转录本组装的过程中存在较多的嵌合体,并且不能准确的得到完整转录本的信息,从而对后面的表达量分析、可变剪接、基因融合等分析造成了较大的影响。基于PacBio的单分子实时测序技术,目前平均读长可达到80Kb,其长度已经超过一般转录组中典型的基因的长度,所以利用长读长测序平台进行转录组的研究,可以直接得到全长转录本信息,而无需组装,从而更大限度的保证了转录组测序结果的准确性。
研究内容
全长转录组基于PacBio的单分子实时测序技术,可以直接得到全长转录本信息,无需组装,更大限度的保证了转录组测序结果的准确性,补充已注释基因组的基因注释结果,发现新的基因和转录本,鉴定可变剪接、基因融合现象、APA位点(Alternative polyadenylation,选择性多聚腺苷化位点)等,并改善基因表达定量。
对于无参考序列的物种,全长转录组测序可以构建高质量的基因集,为后续物种的功能研究奠定基础。
对于有参考序列的物种,可发现新的转录本,完善参考基因集;并鉴定可变剪接、基因融合等结构变异。
此外在基因组研究中,全长转录组还可以用于辅助基因组基因注释
产品优势
华大特有技术:多倍体通量全长转录组和polyA全长转录组是华大特有技术。利用UMI技术可以实现准确定量;多倍通量相同数据量可以获得3-5倍的有效reads,检测转录本数量翻倍;polyA全长转录组可以获得全长polyA尾的长度,可研究其生物学功能。
策略多样:除常规的全长转录组文库外,还可根据需求增建5-10K大片段文库,满足客户对长转录本的研究需求。
通量大:华大基因拥有12台Sequel测序仪,通量大,测序成本低,周期短。
样品起始量低:华大基因全长转录组样本需求仅1ug。
信息分析内容全面:实时跟进科学研究前沿,不断升级信息分析内容。
个性化分析:具有丰富个性化分析经验,可根据项目需要选择更适宜的分析软件,只为保障更精准结果。
无需组装:长读长不需要组装,就可以准确的得到全长转录本的序列信息。
精准基因集:借助长读长测序平台的优势获得更精准基因集,可以改善基因表达定量的结果。
更多新发现:可以发现新的基因和转录异构体,并准确的鉴定可变剪接及基因融合现象。
辅助基因注释:可辅助基因组de novo基因注释,获得更好的基因注释结果。
经验丰富:华大基因自2015年推出全长转录组产品以来,已完成上千个全长转录组测序。目前华大基因的Sequel平台运行良好,实验及信息分析人员上机及问题处理经验丰富。
案例一:三代测序研究玉米转录组的复杂性
Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing(Nature communications, 2016).
方案设计:
材料选择:玉米自交系B73不同发育阶段的6个组织(根、花粉、胚芽、胚乳、幼雌穗、幼雄穗),提取RNA;
测序策略:
二代测序:6个组织进行二代RNA-Seq测序,每个样品三个重复;
三代测序:每个样本反转录之前加入特异性barcode,后续进行等量混合,上机测序47cell;
分析方案:检测玉米可变剪接现象;转录因子分析;lncRNA分析;融合基因分析;甲基化分析。
主要结论:
- 构建5种不同片段大小的文库,上机测序47cell,总共产生3,716,604条reads,过滤掉低质量的reads,总共获得1,553,692
条全长的转录本序列(FL)。
- 和RefGen-V3的isoforms进行长度比较,发现全长转录本预测出来的转录本整体上比V3基因集的要长。在目前的V3基因集中一个基因平均有2.84个isoforms,而全长转录组数据显示,一个基因平均有6.56个isoforms,是前者的两倍多。Isoforms的组织特异性分析显示:花粉有更高的组织特异性,而根的特异性最低。
- 在玉米的V3参考基因集中,转录因子数目为2,624,分为57个家族。全长转录组解决方案将转录因子的数据增加到5,423个,几乎是两倍
- 鉴定出878个lncRNA,其中11个是以前报道过的,867个是新的lncRNA。 这些lncRNA的平均长度为1.1kb(范围为0.2kb-6.6kb),比之前的认知的lncRNA要长很多(平均400+kb)。花粉拥有最多的特异的lnc(238个),穗是最少的lnc(68)个。
- 从Pacbio数据中鉴定出1,430个融合转录本。其中143个被Illumina数据支持。结果表明,融合事件多发生在染色体间。
案例二:三代和二代测序结合研究揭示丹参药用成分合成机理
Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms. (Plant Journal,2015)
方案设计:
研究材料:丹参酮一般认为产生于丹参根部周皮部,研究分别取了根部的周皮(periderm)、韧皮(phloem)、木质(xylem)3种类型的根部组织进行了mRNA测序。
研究方法:3种类型根部样本各设置3个生物学重复,总共9个样本,采用HiSeq2500 PE100进行测序,每个样本产生~5G raw data 。9个样本混合测序,采用PacBio RSII 进行测序,建<1kb、1-2kb、2-3kb、>3kb 四个SMRT bell文库,总共产生~4.8G raw data;
主要结论:
- 采用HiSeq2500 数据对PacBio RSII平台所产生的subreads进行了校正,最后得到了16,241个高质量非冗余isoforms。
- 基于HiSeq2500产生的mRNA数据的差异表达分析,发现了在根部周皮部特异表达与者高表达丹参酮合成相关基因,SmCPS1、SmKSL1、GGPS、IPI、CYP等;
- 最后研究者使用得到的16,241个高质量的Isoforms进行了可变剪接分析,发现了大约有40%检测基因位点发生了可变剪接现象,其中有些基因参与了萜类化合物代谢及类异戊二烯代谢。
多倍通量全长转录组
为了充分利用sequel测序越来越多的数据,华大基因Dr.Tom主导研发了多倍通量全长转录组产品,在建库过程中将多个转录本首尾相连构建sequel文库;通过CCS测序,一条CCS read可得到多条转录本,大幅提升sequel测序全长reads的获得率。
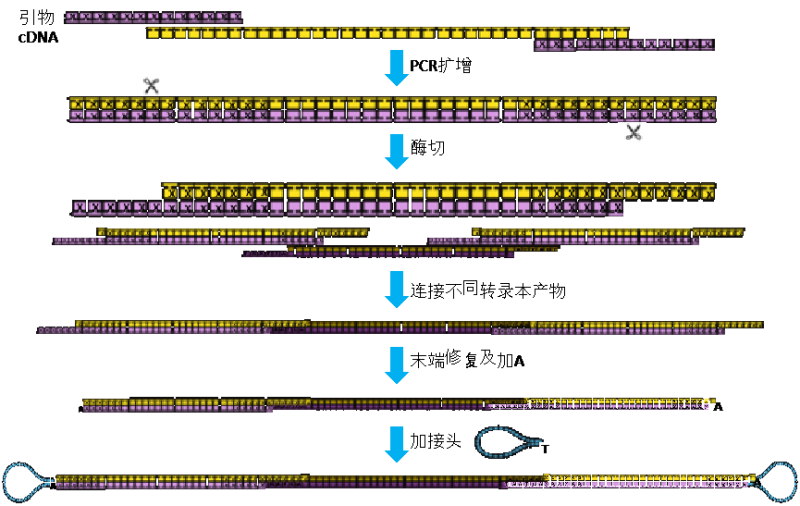
图1 多倍通量全长转录组建库示意图
多倍通量全长转录组可将单个Sequel cell测序得到的全长reads数提升3-5倍以上;检测到的基因和转录本数目都有明显提升,并结合UMI可实现准确的基因或转录本定量。
1) 全长reads数提升可达3-5倍以上
同样的测序数据量,多倍体通量全长转录组产品测序一个cell检测到的reads数是常规sequel检测方法的3-5倍,最高可达7倍以上。
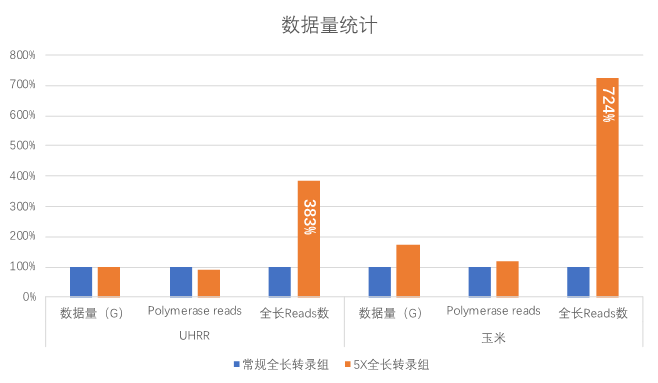
图2 Reads数统计。排除原始数据产量的影响,多倍通量全长转录组测序得到的全长reads数可高达常规文库的7倍以上。
2) 转录本检测数目可提升1倍以上
多倍通量全长转录组产品1个sequel cell检测到的基因和转录本数目远高于常规文库,转录本数目检测数目可提升1倍以上。
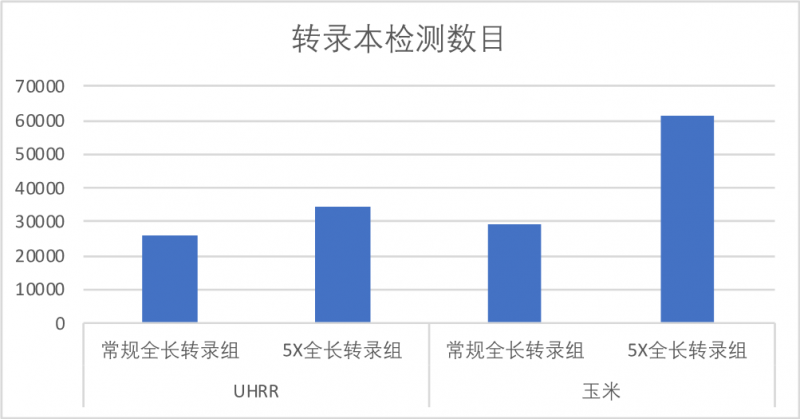
图3 转录本检测数目。多倍通量全长转录组产品测序1个cell检测到转录本数目可提升1倍以上。
3) 基因定量结果更准确
多倍通量全长转录组产品将UMI(Unique molecular identifier,特异性分子标识符)和数据量提升完美结合,全长转录组也可以做基因定量。如下图所示,多倍通量全长转录组基因定量结果和短读长测序(PE测序)定量结果一致性好。

图4 基因定量一致性(spearman系数)
4)选择多倍通量全长转录组产品,大部分物种测20G数据就可满足研究需求
有了多倍通量全长转录组产品,对于转录本复杂度不是太高的物种,一般测20G数据就可满足研究需求。
polyA全长转录组
大多数真核生物的mRNA 3’末端都有由几十个到几百个不等的A碱基组成的Poly(A)尾巴。在目前的mRNA测序中,它们的信息一直被忽略。事实上,它们对于mRNA的稳定性和蛋白翻译非常重要,一些常见生理生化途径可能都会受到Poly(A)长度的调控作用。常规的转录组测序中,Poly(A)只是作为反转录引物结合位点,反转录后只保留约20个Poly(A)碱基,Poly(A)本身的信息一直是被忽略的。利用polyA全长转录组测序可以获得全长polyA尾序列,基于此可以进行polyA长度相关的研究。

图5 polyA全长转录组建库方法示意图
1)polyA长度分布:PolyA长度从几十个碱基到几百个碱基不等。细胞不同发育时期,转录本polyA长度不同
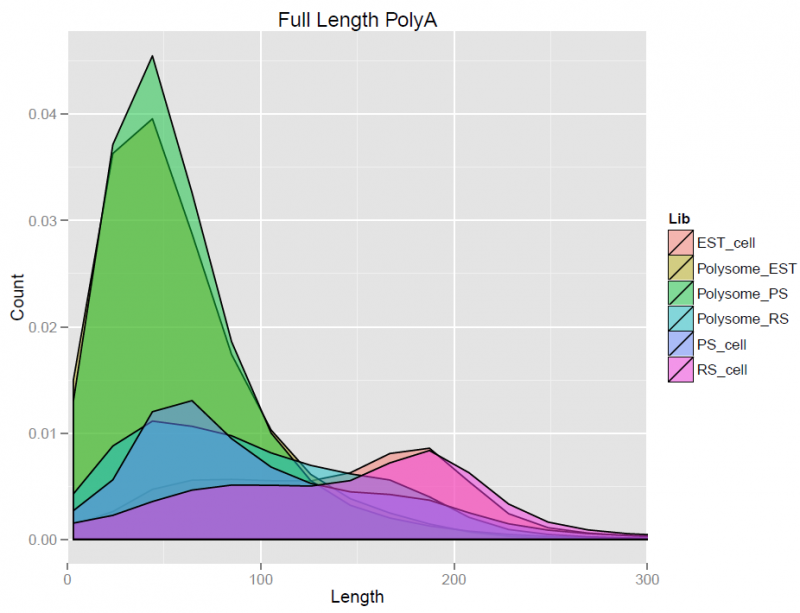
图6 polyA长度分布
2)polyA中其他碱基含量:polyA主要是由A碱基组成,也会包含一些其他的非A碱基,如C, G, U等。这些非A碱基可能会影响polyA相关的功能。

图7 polyA中其他碱基含量
3)polyA 长度与可变剪接及转录本表达量关系
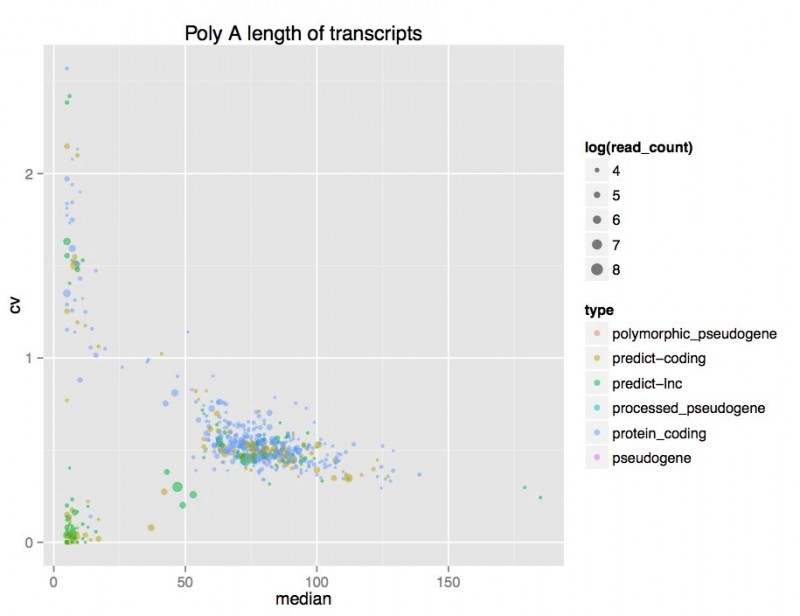
图8 polyA 长度与可变剪接及转录本表达量的关系
RNA样本送样建议
| PacBio转录组(Sequel) | ||||||
| 样本类型 | 总量 | 浓度 | RIN | 28S/18S | 基线和5S | 纯度 |
| Total RNA | 1μg | 285ng/μl | RIN≥8.0 | 28S/18S≥1.4 | 基线平整,5S峰正常 | OD260/280≥1.8 |
| OD260/230≥1.8 | ||||||
组织样本送样建议
|
组织类型 |
送样量(提取RNA) |
|
新鲜动物组织干重 |
50-200mg |
|
新鲜植物组织干重 |
500-1000mg |
|
新鲜培养细胞 |
5×107--2×108个 |
|
全血(哺乳动物) |
2-5mL |
|
全血(非哺乳动物) |
0.5-2mL |
|
菌体 |
2×107--2×108个 |
|
藻类 |
2×107--2×108个或1-2g |
Q1:PacBio平台的测序原理?
A1:PacBio是基于单分子,实时测序Single-Molecule, Real-Time(SMRT)技术:建好的文库放在SMRT Cell上进行测序,Sequel SMRT cell中有100万个ZMW;每个孔下面固定DNA合成酶,当待测DNA分子下降到孔中时会与DNA合成酶结合,同时在该酶的催化下,进行了DNA链的合成。由于使用了带有荧光标记的dNTP,在合成时荧光基团会发出亮光,通过检测亮光来读取碱基。
Q2:PacBio平台转录组产品的优势?
A2:相比于HiSeq平台的转录组产品,PacBio转录组不需要组装,就可以得到全长转录本的信息。而二代测序由于读长的限制,得到的全长转录本的数量、准确度及完整性是大打折扣的。
Q3:PacBio目前能接原核转录组吗?
A3:目前全长转录组主要针对真核转录组,如果需要做原核转录组或LncRNA,可以个性化沟通。
Q4:Sequel转录组还需要建3-4个文库吗?
A4:之前用RSII测序平台时推荐建3-4个文库,主要是因为RSII上机偏向性较大,如果要是构建1个文库,而文库的片段范围是1-10K的话,那上机的时候的数据会有大部分都是小片段的,大片段的转录组所占的比例会非常少,数据量也就很少,因为小片段更利于掉入ZMW小孔中。
Sequel平台在上样偏向性上有较大的提升,不需要构建3-4个文库,构建一个文库(1+0.4X磁珠纯化文库)即可得到和转录本实际分布相符的结果;若物种长转录本较多,也可以增建一个4.5-10k文库。
Q5: PacBio Sequel转录组推荐的测序方案?
A5:建库:0-5kb文库(1+0.4X磁珠纯化文库);测序:1-2个Cell。
转录组0-5K文库也包含5K以上片段,测序结果反映转录本的真实情况。
如果特别关注5K以上的长转录本,也可以增加一个4.5-10K的文库。
Q6:全长转录组结题报告中的数据量怎么看?
A6:关于全长转录组结题报告数据量解读,目前全长转录组结题报告中有下机数据量统计,subreads统计以及reads of insert(CCS)统计。Polymerase, subreads 和reads of insert(CCS)对应关系如图1所示:
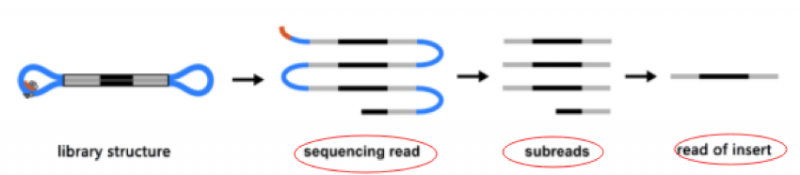
图1 全长转录组测序结果展示图
表1是下机数据统计,就是我们一般承诺的数据量,这里选择了一个数据量小的项目(或者是截取了一部分数据)做的流程。Total base就是下机数据量。Total reads一般跟P1的比例有关(~1,000,000*P1,1,000,000是Sequel的ZMW孔数)。平均读长代表的酶读长,Sequel一般承诺8K以上,读长越长,CCS序列的准确性越高。
注意:三代下机数据是自动过滤了质量值小于0.8和读长小于50bp的片段后的数据,不需要像二代测序那样再进行数据过滤,所以三代下机的都是有效数据,没有clean data和raw data的概念。
表1 聚合酶读取(Polymerase Reads)情况汇总
|
Sample |
Library |
Cell Number |
Total Reads |
Total Base(GB) |
MaxLength (bp) |
MeanLength (bp) |
N50 Length(bp) |
|
1 |
A |
1 |
494927 |
8.78 |
113085 |
17737.35 |
35855 |
表2是subreads数据统计,代表原始下机数据去了接头以后的数据。一条polymerase去接头以后会生成多条subreads,所以这里的total reads数比较多。总数据量和去接头前没有大的差别。平均长度跟插入片段长度有关。这个平均长度比表3的平均长度短,可能是受一些不完整片段的长度影响。
表2 Subreads情况汇总
|
Sample |
Library |
Cell Number |
Total Reads |
Total Base(GB) |
MaxLength (bp) |
MeanLength (bp) |
N50 Length(bp) |
|
1 |
A |
1 |
3836208 |
8.78 |
72300 |
2288.38 |
4661 |
表3 测序情况汇总
|
Sample |
Library |
Cell Number |
Reads of Insert |
Read Bases of Insert(bp) |
Mean Read Length of Insert(bp) |
Mean Read Quality of Insert |
Mean Number of Passes |
|
1 |
A |
1 |
484946 |
1,758,449,481 |
3626 |
0.9 |
6 |
表3是CCS数据统计,就是之前结题报告中提供的数据。对表2中每条序列的多个subreads进行自纠错,得到一条read of insert,read of insert的数量是和表7的total reads数基本一致的(略偏少)。平均长度跟转录本的平均长度接近。