- 首页 > 动植物全基因组重测序
动植物全基因组重测序
全基因组重测序是对已知基因组序列的物种进行DNA测序,并在此基础上完成个体或群体分析。全基因组重测序通过序列比对,可以检测到大量变异信息,包括单核苷酸多态性(SNP)、插入缺失(InDel)、结构变异(SV)和拷贝数变异(CNV)等。基于检测到的变异能进一步研究动植物的物种特性、群体进化问题、定位目标性状基因位点。
随着测序成本降低和已知基因组序列物种的增多,全基因组重测序已经成为动植物分子育种、群体进化中最为迅速有效的方法之一。利用全基因组重测序技术有助于快速发现与动植物重要性状相关的遗传变异,应用于分子育种中,缩短育种周期。
产品优势
· 技术简单,稳定性好。
· 检测变异类型丰富:可以检测SNP、InDel、SV和CNV等多种变异类型,并可用作分子标记。
· 高密度标记: 能够检测到全基因组范围的SNP信息,同时可检测低频SNP。
· 发现新的变异:与芯片方法相比较,可以检测到新的变异序列。
· 高性价比:与全基因组从头测序相比,耗时更短,成本更低。
· 样品起始量低:华大基因经过不断的研发,样本起始量不断降低,最低可至pg级。
· 个性化分析:具有丰富个性化分析经验,可根据项目需要选择最适宜的分析软件,只为保障最精准结果。
· 数据精准:华大至今完成10万+的动植物重测序样本,严格质量控制流程保证结果准确度。
· 经验丰富:动植物重测序领域挂名发表文章100余篇,IF加和>1,000,其中一作或通讯作者文章50+,涵盖变异检测、遗传图谱构建&QTL定位、群体进化和GWAS等各研究领域。
· 项目方案支持:大项目参与方案设计,使项目赢在起跑线。
· 分析团队实力雄厚:发表影响因子10分以上动植物研究文章的人员20+。
信息分析内容
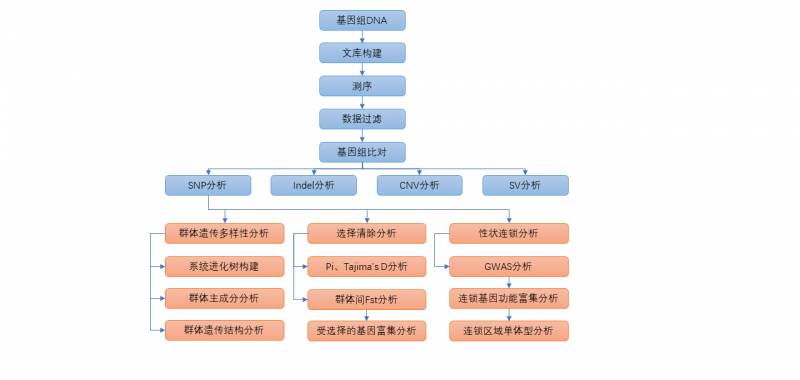
产品应用

案例一、群体研究案例——322个不同的红豆品种基因组变异图谱的绘制揭示基因组进化与关键的农艺性状
2024年3月,Plant Biotechnology Journal 在线发表了北京农学院联合华大基因,中科院遗传与发育研究所和澳大利亚莫道克大学题为 “Chromosome-level reference genome and resequencing of 322 accessions reveal evolution, genomic imprint and key agronomic traits in adzuki bean” 的研究论文。研究人员开发了红豆品 “Jingnong6” 的高质量染色体级别参考基因组,通过结合 PacBio Sequel长读序列测序以及短读序列和 Hi-C 技术,组装的基因组覆盖了红豆基因组的 97.8%,contig N50 约为16 Mb,总共包含 32,738 个蛋白质编码基因。此外,通过全基因组重测序(WGRS)技术对 322 个不同的红豆品种进行了基因组变异图谱的绘制,包括野生和栽培品种。通过比较基因组学和全基因组关联研究(GWAS),研究人员鉴定了几个与驯化性状显著相关的候选基因,包括VaCycA3;1、VaHB15、VaANR1 和 VaBm。特别是,VaANR1 和 VaBm 在调节种皮颜色方面的作用进行了功能性分析。该研究为赤豆育种工作提供了基因组数据,并揭示了赤豆驯化性状的关键基因。研究为育种工作提供了重要的数据基础,有助于改良赤豆的耐逆性和产量等关键农艺性状,进一步选育拥有良好农艺性状的高品质赤豆品种。
研究策略:红豆进行基因组从头组装,并对322个红豆品种进行全基因组重测序,包括 67 个野生种、39 个半野生种、160 个地方品种和 56 个改良品种,平均基因组覆盖度为 8.92X。
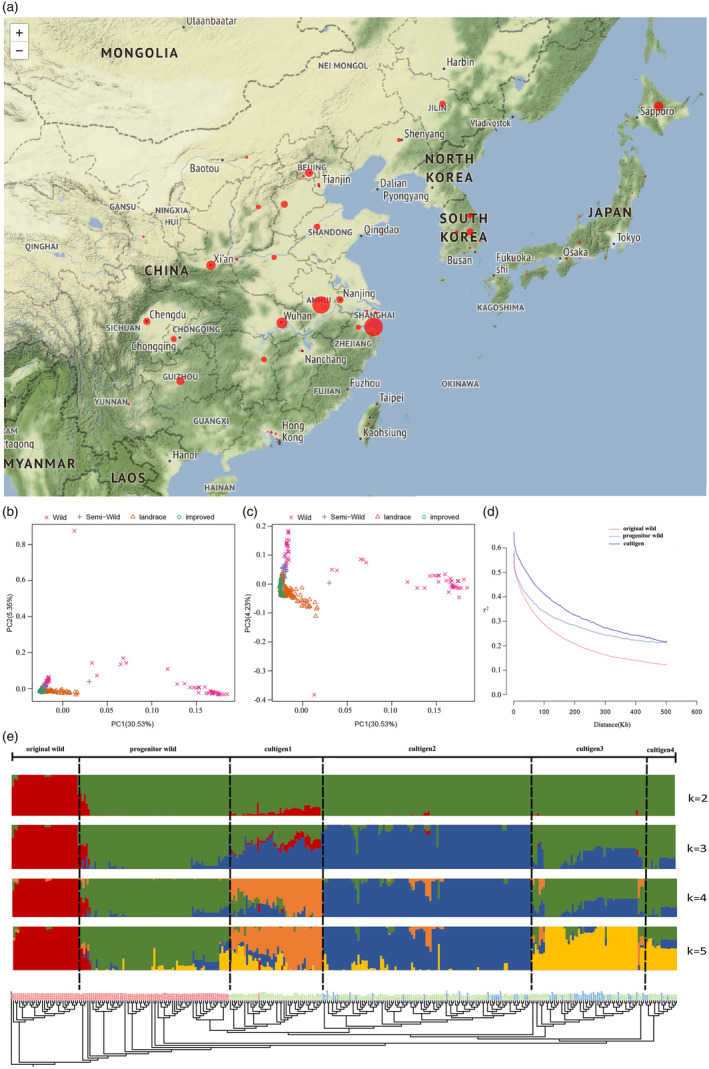
图1 322份野生和栽培红豆的种群结构
案例二、DNBSEQ群体重测序——生菜驯化历史及GWAS分析
华大与荷兰遗传资源中心、深圳国家基因库、华中农业大学等多家单位合作,在Nature Genetics杂志发表题为“Whole-genome resequencing of 445 Lactuca accessions reveals the domestication history of cultivated lettuce”的研究论文,对来自全球47个国家的445份生菜种质资源利用DNBSEQ平台进行测序,除12个野生种50X以上进行基因组组装,其他样本为20X重测序,囊括了生菜的所有栽培类型及主要野生近缘育种材料。全面揭示了栽培生菜的完整驯化进程,并对生菜的种质资源结构、重要农艺性状和抗病基因来源进行了探索研究。
研究团队通过系统进化分析发现,所有生菜样品在进化树上聚为一支,与野生近缘种野莴苣(L. serriola)有着最近的共同祖先,而且所有栽培生菜都源自一次独立的驯化事件。对主成分分析和群体结构进行解析,我们将野莴苣分为六个具有不同群体特征的地理居群,而栽培生菜与高加索地区、两河流域的野莴苣居群具有最近的遗传距离。由此推断,栽培生菜极有可能起源于高加索地区、两河流域。

图2 栽培生菜(图中绿色所示)与野生近缘种的群体分析
通过有效群体大小分析发现,距今1万年栽培生菜和野莴苣均经历了种群收缩,可能由环境剧烈变化所致。而从公元前4000年开始,栽培生菜有效群体大小出现了更为剧烈的下降,暗示着生菜正在经历人工驯化。对生菜种群结构和发展趋势进行深入的分析,推测生菜最早在高加索或近高加索地区被驯化。在被人类驯化之后,生菜先传播到古埃及并逐渐演变为如今的油用生菜。在古罗马时代传到南欧地区,与当地的野莴苣杂交之后,开始作为叶用生菜种植食用。

图3 栽培生菜起源中心推测近高加索地区
栽培生菜有很多所谓的 “驯化综合症(domestication syndrome)”,如叶片全缘、缺少叶刺、无法散种等。本研究通过全基因组关联分析,对重要的驯化和农艺性状相关基因进行了精细定位。将以上三个驯化性状的相关基因座位,分别定位于生菜基因组的第 3、5 和 6 号染色体上。对散种基因所在区域的变异位点进行系统进化分析,发现栽培生菜与高加索的野莴苣居群在进化树上最近,揭示了散种的丢失可能是发生在生菜驯化的早期事件。对全缘叶关联区域进行系统进化分析,发现栽培生菜与南欧的野莴苣居群聚为一支,表明全缘叶这一性状很可能来自南欧地区的野莴苣。

图4 生菜基因组中受人工选择区域与重要驯化性状关联区域
叶用生菜在种植期间,易受各种病虫害侵扰,其中由莴苣盘梗霉(Bremia lactucae)导致的霜霉病最为严重,在生菜生长期均可发病。成株期的叶片发病影响生菜外观品质,严重时损失可达 20-40%,所以从野莴苣中鉴定霜霉病抗病基因一直是生菜育种中的重要内容。为了挖掘生菜基因组中的抗病基因资源,对栽培生菜和野莴苣进行了比较基因组分析,发现位于第 1、2 和 4 号染色体的主要抗病基因簇有着更多的野莴苣基因渗入。本研究利用霜霉病小种抗性调查数据开展全基因组关联分析,发现栽培生菜的抗性位点通常位于单一抗病基因簇,而野莴苣的抗性基因座位则分布在不同染色体上,这表明利用野莴苣开展抗病育种有非常大的价值。

图5 野莴苣基因渗入(红色线条)对生菜基因组主要抗病基因簇的贡献
案例三、群体研究案例——3K水稻重测序&泛基因组研究
由中国农业科学院作物科学研究所牵头,联合 IRRI、上海交大、华大基因、深圳农业基因组研究所、安徽农大等 16 家单位共同完成了“3000份亚洲栽培稻基因组研究” ,并于 2018 年 4 月发表在 Nature 上。研究针对水稻起源、分类和驯化规律进行了深入探讨,揭示了亚洲栽培稻的起源和群体基因组变异结构,剖析了水稻核心种质资源的基因组遗传多样性。
3000 份水稻(来自全球 89 个国家和地区)代表了全球 78 万份水稻种质约 95% 多样性的核心种质。通过全基因重测序,每个样本平均测序深度14X,利用重测序数据共检测到 32M 的高质量 SNPs 和 InDels。对亚洲栽培稻群体的结构和分化进行了更为细致和准确的描述和划分,由传统的 5 个群体增加到 9 个。研究着重分析了 453 个测序深度 >20X 品系的 SVs,利用 SVs 构建的进化树与 SNP 构建的进化树类似。大量的 SVs 可能是不同程度杂种不育和 XI 与 GJ 杂种衰退的遗传基础。同时构建了亚洲栽培稻的泛基因组,包括 12,770 个(62.1%)核心(core)基因家族和 9,050 个(37.9%)分散式 (distributed) 基因家族。发现了 1.2 万个全长新基因和数千个不完整的新基因。核心基因比较古老,大多数的新基因表现更年轻和长度偏短。

图6 水稻泛基因组研究
a、基因家族PAVs;
b、泛基因组和一个单独的基因组的组成成份;
c、基于500个随机筛选的水稻基因组模拟泛基因组和核心基因组;
d、核心和分散式基因家族比例;
e、两个品系间基因家族平均数量差异;
f、5733主要群组不平衡基因家族特性
变异检测
全基因组重测序数据与参考基因组比对,可以检测某物种个体或群体的遗传变异信息,包括单核苷酸多态性(SNP)、插入缺失(InDel)、结构变异(SV)、拷贝数变异(CNV)。变异信息是进行其他信息分析的基础。

图1 各种变异在基因组上分布统计
从外到内依次为:染色体坐标、SNP密度分布、InDel密度分布、SV类型分布、CNV的拷贝数分布。
群体结构分析
通过构建群体的系统进化树(图2a)、主成分分析(图2b)和Structure分析(图2c),研究样本间的亲缘关系和进化关系。进化树是根据样本间亲缘关系的远近,把各样本安置在有分枝的树状的图表上,简明地表示生物的进化历程和亲缘关系。主成分分析(Principal Component Analysis,PCA),是将多个变量通过线性变换以选出较少个数重要变量的一种多元统计分析方法。群体结构研究的过程中通过将测序品系和SNP位点构成二维矩阵数据,经过PCA分析,计算出几个主要的特征向量,并且将每一个品系在各特征向量上进行定位,也是研究群体品系间亲缘关系的方法之一。Structure分析则是假设若干个品系起源于K个截然不同(或差异较大)的祖先,分析每一个品系的遗传成分中,所具有的每一个假想祖先成分的比例。三种分析方法的结果可以相互验证。

图2 群体结构分析( Nature genetics, 2010, 42(12): 1053-1059)。
a为进化树;b为PCA分析;c为Structure分析,不同颜色代表不同的假想祖先;d为连锁不平衡分析
连锁不平衡分析
连锁不平衡(linkage disequilibrium,LD),指群体内不同座位等位基因之间的非随机关联, 包括两个标记间或两个基因间或一个基因与一个标记座位间的非随机关联,可以用r2计算两个标记间的连锁不平衡度。LD受重组、人工选择、群体类型等的影响,不同的物种LD变化情况不同,一般情况下我们会统计LD值衰减到一半的距离(图2d)。LD值会对信息分析中标记数目的选择有指导意义,LD大的物种所需要的标记密度相对低。
选择分析(条件:群体有明显的亚群分化)
选择在物种的遗传变异形成过程中有巨大的贡献,其中搭便车效应会对种群水平的分化产生剧烈的影响,由于较强的选择效应,使得一个突变位点相邻DNA上的核苷酸之间的差异下降或消除(selective
sweep)。通过分析大量的比较基因组学数据集和大量的SNP集,我们可以确定在野生种到栽培种/地方种的过程中,以及在不同的环境情况下,哪些区域的多态性发生了巨大的改变,检测驯化或环境适应性相关的候选基因,而且受选择的基因与进化相关的性状也有关系。

图3 选择分析结果示例( BMC plant biology, 2015, 15(1): 81)
绿色区域代表栽培种驯化过程中受选择区域
GWAS分析
利用分布于全基因组水平的分子标记(例如SNP)通过一定的模型(如一般线性模型或混合线性模型)与表型进行关联分析,检测目标性状相关基因位点。但是由于连锁的存在,往往我们检测到的标记并不是直接决定目标性状的变异,如果进行基因克隆时还是要在一定的定位区间内完成。

图4 GWAS结果示例(Nature genetics, 2010, 42(11): 961-967)。
Manhattan plot(图4左)和QQ plot(图4右)是查看GWAS定位结果和计算模型合理性的标配图。Manhattan plot横坐标是表示位置,纵坐标表示-lgP,在纵坐标上超过一定阈值的点被认为和表型关联。QQ图的意义在于基因型和性状无关联的情况下,各个标记P-value的观察值和期望值是相等的(红线),但是由于出现了基因型和性状有关联的情况,P-value往往会偏离y=x这条线。
表1 基因组 DNA样品送样建议
样本类型 | 总量 | 浓度 | 完整性(胶图) | 纯度 | |
Genomic DNA | 常规文库 | ≥0.2 μg(推荐 0.4 μg) | ≥8 ng/μL | 主峰>20Kb | 无蛋白, RNA/盐离子等污染, 样本无色透 |
PCR free 文库 | ≥1 μg(推荐 2 μg) | ≥12.5 ng/μL | 主峰>20Kb | ||
表2 组织样品判定标准
组织类型 | 常规DNA小片段文库 | PCR free文库 |
新鲜培养细胞 (细胞数) | ≥5×106 cells | ≥5×106 cells |
新鲜动物组织干重 | ≥25 mg | ≥50 mg |
新鲜植物组织干重 | ≥200 mg | ≥200 mg |
全血(哺乳动物) | ≥0.6 mL | ≥0.6 mL |
全血(非哺乳动物) | ≥0.1 mL | ≥0.1 mL |
Q1: 进行全基因组重测序数据推荐?
答:每个样本推荐的数据量与样本类型和要做的信息分析内容相关。例如关注个体样本的SNP,对SNP的准确度和覆盖度要求比较高,一般推荐测序深度>30X,对于稀有变异测序深度还要进一步提高;用于研究群体结构的样本,测序深度推荐10X以上;纯合样本混样检测等位基因频率,推荐平均每个样本的测序深度在1X以上,混合样本测序深度不低于30X;DH和RIL群体构建Bin Map,子代群体测序深度可以测序1X/样本。
Q2: 样本量选择多大合适?
答:样本量大小与样本类型和研究目的相关。例如进行群体进化研究推荐30个样本以上,因为从统计学上说30个以上才属于大样本;对于进行基因挖掘的项目来说,无论是利用自然群体进行GWAS分析或是用家系群体进行连锁分析,都是群体越大越好,一般的情况下进行GWAS分析的样本推荐300个样本以上,对于家系群体推荐200个以上。
Q3: 连锁图谱构建适用于什么样的群体?
答:连锁图谱的构建适用于作图群体,它是由性状差异显著的亲本杂交衍生的群体。亲本选择的要求:要考虑亲本间的遗传多态性、目标性状差异、亲本的纯合度和杂交后代的可育性。构建分离群体类型,根据遗传稳定性可将分离群体分成两大类:暂时性分离群体如F1、F2、BC等,永久性分离群体如RIL、DH等。
Q4: 重测序reads与参考基因组比对率低,可能的原因是什么?
答:重测序reads比对率低原因可能是:1)因为测序样本与参考基因组亲缘关系比较远。因为动植物品种多样,但是目前已完成基因组组装的往往只是其中的一个品种,同一个物种野生种与驯化种差异还是很大的;2)可能因为DNA不纯,存在其他物种的污染;3)参考基因组序列组装质量较差,引起比对率低;4)比对参数设置严格等