- 首页 > ChIP-Seq
ChIP-Seq
染色质免疫共沉淀(ChIP)是在体内环境中研究蛋白质与DNA相互作用的经典实验方法,广泛应用于组蛋白修饰、特定转录因子的基因调控作用等相关领域。随着新一代测序技术的发展和成熟,染色质免疫沉淀实验与高通量测序的整合——Chromatin Immunoprecipitation Sequencing (ChIP Sequencing ),可在全基因组范围对蛋白结合位点进行高效而准确的筛选与鉴定,同时也为研究的深入开展打下基础。
采用特异性抗体对目的蛋白进行免疫沉淀后,分离与其结合的基因组DNA片段,再通过高通量测序与数据分析,在全基因组范围内寻找目的蛋白的DNA结合位点,并且可以基于多个样品进行差异比较。
技术流程

技术优势
数据利用率高:SE50 最优读长,无数据浪费
低起始量:DNBSEQ 平台最低起始量仅需 5 ng,5 ng 以下亦可做定制化
交付周期短:30 个工作日交付结果(不包括 ChIP 实验),DNBSEQ 平台最快交付周期仅需 20 个工作日
解决方案咨询:根据客户实际情况提供相关解决方案
性价比高:基于抗体富集目标区域,有效降低测序数据量
测序范围广:全基因组范围内的DNA与蛋白相互作用进行测序分析
关联分析:可定制 ChIP-Seq 与 RNA-Seq 差异表达基因(DEGs)关联分析
信息分析
表1 标准信息分析内容
信息分析条款 | 信息分析内容 |
标准信息分析 | 1) 数据基本处理与质控; 2) 基因组测序深度累积分布; 3) Peak 扫描 4) Peak 长度分布 5) Peak 深度分布 6) Peak 在基因功能元件上的分布 7) Peak 相关基因分析 8) Peak 相关基因的 GO 功能显著性富集分析 9) Peak 相关基因的 Pathway 功能显著性富集分析 10) 鉴定样品间差异 Peak 11) 样品间差异peak在基因功能元件的分布 12) 样品间差异 peak相关基因 13) 样品间差异peak 相关基因的 GO 和KEGG富集分析 14) Motif 分析 |
定制化信息分析 | 可结合客户的需求,协商确定定制化信息分析内容 |
案例一、DNBSEQ 平台 ChIP-Seq在有丝分裂期间染色质结构动态变化的研究 [1]
案例描述:
在本研究中探索了组蛋白修饰与有丝分裂中的染色质区室化之间的相关性。通过染色质免疫沉淀测序(ChIP-seq)技术,研究人员对缺乏凝缩蛋白的有丝分裂细胞中的活性染色质标记进行了分析,包括 PolII、H3K27ac、H3K36me3 和 H3K4me1。研究发现,在没有凝缩蛋白的情况下,有丝分裂染色体上完全没有PolII的存在,排除了转录作为有丝分裂区室化驱动力的可能性。在 mA1 区室中,H3K27ac、H3K36me3 和 H3K4me1 的富集程度高于 mA2 区室。特别值得注意的是,与其它两种标记相比,H3K27ac 在 mA1 和 mA2 之间的差异更为显著,这表明 H3K27ac 信号强度的差异可能与 mA1 和 mA2 在有丝分裂期间不同的相互作用偏好性有关。
发表单位:深圳湾实验室
发表期刊:Nature Genetics
影响因子:29.0
研究技术:(DNBSEQ ChIP-Seq)染色质免疫共沉淀测序、Hi-C、CUT&Tag 等。
研究成果:
揭示了有丝分裂染色体在缺乏凝聚素时的基因组折叠原则;
发现了有丝分裂染色体在缺乏凝聚素时的新型区室化模式;
为理解基因组在细胞周期中的动态变化提供了新的视角。
部分研究结果展示:
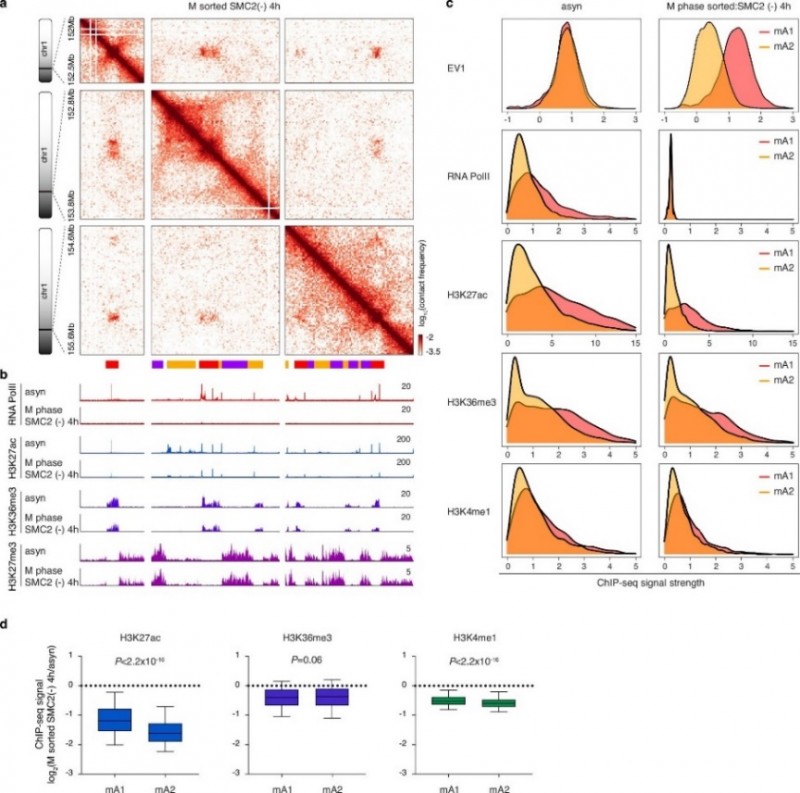
图1 ChIP-Seq 分析 RNA PolII、H3K27ac、H3K36me3 和 H3K27me3 在不同状态的有丝分裂细胞中分布情况
案例二、DNBSEQ 平台 ChIP-Seq 在植物盐胁迫研究中的应用[2]
案例描述:
盐胁迫是限制植物生长和农业生产的一个主要环境因素,全世界超过8亿公顷的土地和20%的灌溉土地受土壤盐分的影响。甲基乙二醛(methylglyoxal, MG)是动物和植物中高度保守的糖酵解副产物,在哺乳动物不同发育过程和疾病、植物对各种胁迫(包括盐胁迫)的反应中发挥作用。盐胁迫增加了甲基乙二醛(MG)的积累,但目前尚不清楚MG调控的基因表达是否与表观遗传修饰有关。
发表单位:武汉大学
发表期刊:Nucleic Acids Research
影响因子:16.6
研究技术:(DNBSEQ ChIP-Seq)染色质免疫共沉淀测序、(DNBSEQ)RNA-Seq、MNase 染色质可及性检测等。
研究成果:
MG调节盐胁迫应答基因的表达;
MG修饰组蛋白H3;
H3甲基乙二醛化与基因表达呈正相关;
H3甲基乙二醛化与盐胁迫应答基因表达相关;
转录因子ABI3和MYC2影响盐胁迫反应基因H3甲基乙二醛化的特异性分布。
部分结果展示:
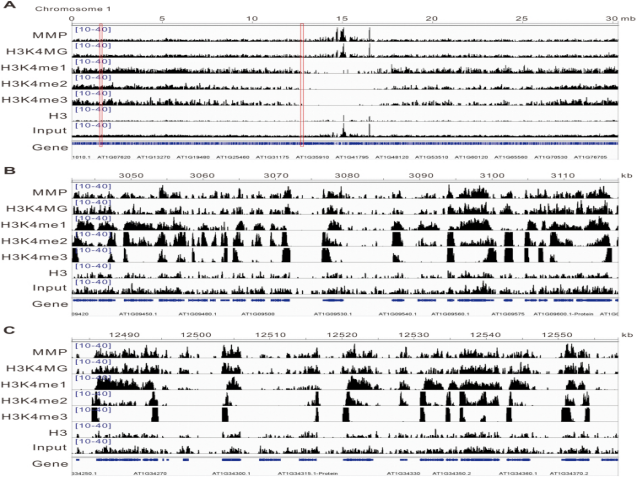
图2 ChIP-Seq分析MMP、H3K4MG、H3K4me1、H3K4me2、 H3K4me3、H3和input
参考文献:
[1] Zhao H, Lin Y, Lin E,et al. Genome folding principles uncovered in condensin-depleted mitotic chromosomes. Nat Genet. 2024 Jun;56(6):1213-1224.
[2] Zheng-Wei Fu , Jian-Hui Li, et al. The metabolite methylglyoxal-mediated gene expression is associated with histone methylglyoxalation. Nucleic Acids Res. 2021 Feb 26;49(4):1886-1899.
1. 差异 peak 相关基因 GO 富集分析
基因本体论(Gene ontology,GO)是所有物种中最主要的了解基因和基因产物属性的生物信息学分析手段,GO 分析能够用于鉴定基因产物的性能,它包含了三类基因功能信息:细胞组分(Cellular Component),分子功能(Molecular Function)和生物学过程(Biological Process)。为了探讨表观遗传变异在通路和生物学过程中起到的作用,我们对DMR 相关的基因进行了 GO 和 Pathway 分析。
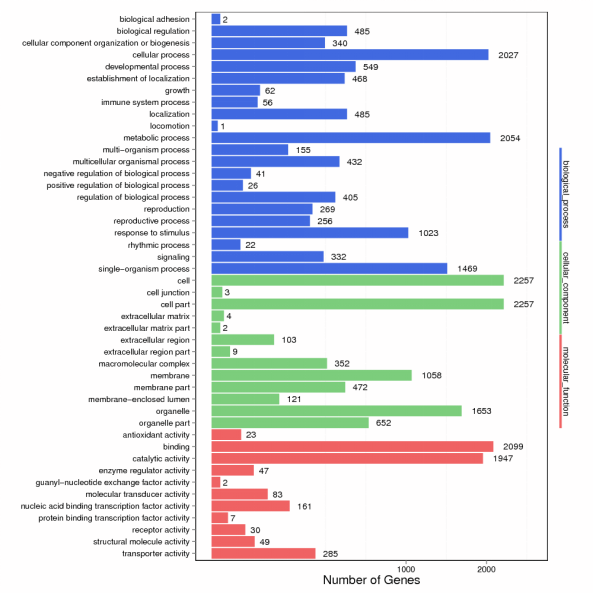
图1 差异 peak 相关基因的 GO 聚类分析
2. 差异 peak 相关基因 Pathway 富集分析
KEGG (Kyoto Encyclopedia of Genes and Genomes) 是有关 Pathway 的主要公共数据库,该数据库整合了基因组、化学以及系统功能信息,特别是测序得到的基因集与细胞、生物体以及生态环境的系统性功能相关联。所有样品中的 DMR 相关基因均用 KEGG 数据库进行分析。
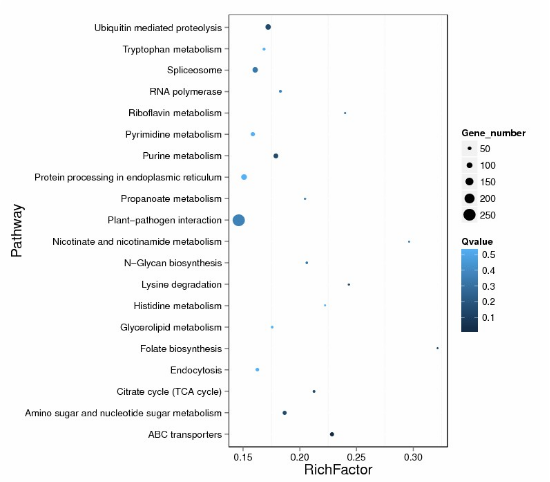
图2 差异 peak 相关基因的 Pathway 功能显著性富集分析
3. 与 RNA-Seq 关联分析
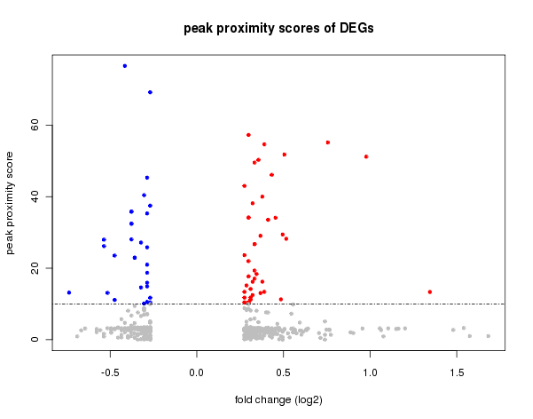
图3 peak proximity scores 与 DEGs 表达差异值关系分布图
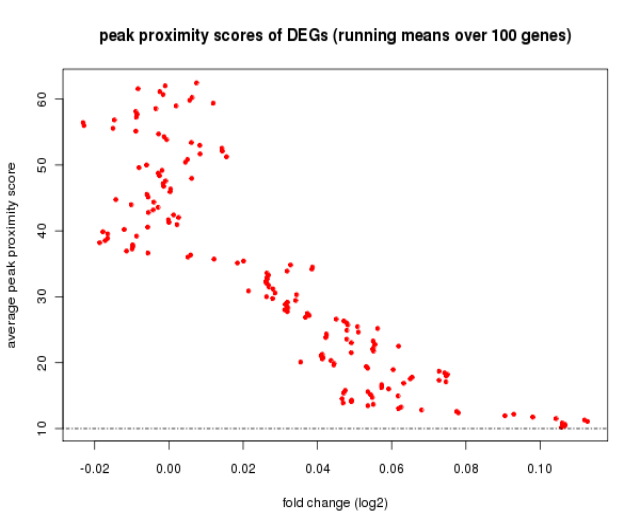
图4 peak proximity scores 与 DEGs 表达差异值趋势图
样品要求
样品类型:IP富集DNA样品,强烈建议同时送对应的Input DNA;
总量:≥ 10 ng ChIPed DNA;
浓度:≥ 1 ng/μL;
纯度:OD260/280= 1.8 -2.0;
DNA片段大小:分布在100~500 bp范围,且主带明显。请提供DNA打断后的检测胶图以确定DNA片段大小是否符合要求,并请附加一份详细的样品信息单并提供ChIP后的q-PCR验证结果;
Q1:DNBSEQ平台的测序原理是什么?
DNBSEQ采用优化的联合探针锚定聚合技术(cPAS)和改进的DNA纳米球(DNB)核心测序技术,是行业领先的高通量测序平台之一。具体而言,首先DNA分子锚和荧光探针在纳米球上进行聚合,随后高分辨率成像系统对光信号进行采集,光信号经过数字化处理后即可获得待测序列。其中,DNB通过线性扩增增强信号,降低单拷贝的错误率。此外,DNB大小与芯片上活性位点的大小相匹配,每个位点结合一个DNA纳米球,在保证测序精度的情况下提高了测序芯片的利用效率。
Q2:DNBSEQ ChIP-Seq产品优势是什么?
a)
准确性高:高深度Peaks与HiSeq平台一致性高达到100%
b) 起始量低:样品起始量低至5ng
c) 周期更短:交付快至20个工作日
d) 性价比高:相同的测序质量,价格更优惠
e) 关联分析:ChIP-Seq与RNA-Seq差异表达基因(DEGs)关联分析
Q3:对样本起始量有没有要求?
建库起始量为5ng,建议ChIP-ed DNA≥ 20ng,低于5ng也可以做,之前有做过无浓度样本成功的案例,但这类项目只能客户自己承担建库风险。
Q4:测序策略是什么?
SE50
Q5: ChIP-Seq的对照中input和IgG有什么不同吗?
Input对照:在进行免疫沉淀前,需要取一部分断裂后的染色质做Input对照。Input是断裂后的基因组DNA,不加抗体做富集,但是需要与沉淀后的样品DNA一起经过逆转交联,DNA纯化,以及最后的PCR或其他方法检测。Input对照不仅可以验证染色质断裂的效果,还可以根据Input中的靶序列的含量以及染色质沉淀中的靶序列的含量,按照取样比例换算出ChIP的效率,所以Input对照是ChIP实验必不可少的步骤。
阴性对照:用普通的IgG做为抗体(目的蛋白抗体宿主的IgG或血清)。理论上不会ChIP下来任何DNA片段,因此作为阴性对照,但是由于非特异结合,或者实验过程中,没发生结合的DNA清除不完全,可能也会出现条带,如果非常明显,那就证明实验过程有待改进。
Q6:推荐多少数据量?能否接不同数据量的项目?
DNBSEQ ChIP-Seq推荐20M clean reads/样本,其他类型数据量也可提供。
Q7:N-ChIP和X-ChIP的区别是什么?
N-ChIP基于核酸内切酶MNase酶切,切割核小体且作用温和,较适用于组蛋白修饰研究;X-ChIP基于甲醛固定,超声打断,适用于大多数蛋白-DNA相互作用研究。
Q8:样品制备过程是否需要PCR扩增?PCR扩增后是否会影响最后的结果?
基于第二代测序平台的ChIP-Seq在样品制备过程中需要进行PCR,但是只需要非常少的反应次数,由于PCR引起的偏向性非常小。另外,在测序结果出来后,针对reads的比对结果,通过信息分析手段可以去除duplication的影响。我们的所有信息分析结果都是在去除duplication之后进行生物学意义挖掘的。根据我们的经验,如果您送的样品是直接ChIP富集的DNA,在我们的样品制备后出现duplication的reads比例不会超过全部reads产量的5%。如果您送的样品在ChIP富集后有做过PCR扩增,那么这一步的PCR对结果的duplication影响非常严重。
Q9:哪些因素会影响ChIP-Seq的结果?
抗体的质量与特异性、需要富集的目标区域在基因组上的比例、ChIP的实验操作、DNA片段长度范围等都会影响ChIP-Seq的结果。