- 首页 > 全长转录组
全长转录组
产品介绍
全长转录组产品,通过长读长测序,无需打断和拼接,直接获取包含 5’UTR,3’UTR 及PolyA 尾的完整转录本,能够全面快速地获得某一物种特定组织或器官在某一状态下的转录本序列及结构信息。可用于补充已注释基因组的基因注释结果、发现新的基因和转录本、鉴定可变剪接、基因融合现象、选择性多聚腺苷化位点等,同时改善基因/转录本表达定量结果,全面解析转录本的复杂性和多样性。
华大科技开发了独特的多倍通量全长转录组产品以及Poly(A)全长转录组产品,提供基因转录本结构、定量,从结果到调控机制的全方位转录调控研究方案。
产品应用
全长转录组基于 PacBio 单分子实时测序技术,可以直接得到全长转录本信息,无需组装,更大限度保证转录组测序结果的准确性。
对于无参考序列的物种,全长转录组测序可以构建高质量的基因集,辅助基因注释,为后续物种的功能研究奠定基础。
对于有参考序列的物种,可发现新的转录本,完善参考基因集;并鉴定可变剪接、基因融合等结构变异。
产品优势
- 多倍通量专利技术
利用华大多倍通量全长转录组专利优势技术,将多条序列随机首尾相连,充分利用长读长测序平台优势。
- 充分利用读长,超高性价比
相同数据量可以获得 3-6 倍的有效 reads,检测转录本数量翻倍。
- Poly(A)+全长转录组,测一得二
同时得到全长 mRNA 序列以及全长 Poly(A) 序列,一举两得。
- Poly(A)长度和碱基检测结果准确度高
基于 PacBio 平台 CCS 测序,可直接测序得到全长的 Poly(A) 长度和碱基信息,准确度高。
- 技术稳定,项目经验丰富
累计执行 1500+ 例样本,涵盖了包括植物、昆虫、哺乳动物、禽类、软体动物和水产生物在内的 240+ 个物种。
- 直接获取全长转录本
直接获得 5’-3’ 的全长 lsoform,无需拼接,所测即所得,Q32-Q36,准确性高。
- UMI 技术实现精准定量
利用 UMI 技术和多倍通量完美结合,基因定量结果更准确。
- 高准确结构分析
提供高精度的 ORF 预测,可变剪接、融合基因分析。
案例一:HIT-ISOseq揭示生菜组织特异性转录组图谱[1]
发表杂志:Communications Biology(IF=5.2)
发表时间: 2024-7-31
方案设计:
- 研究背景:生菜是全球种植和消费最广泛的双子叶蔬菜之一。尽管已有生菜的参考基因组序列,但其基因注释仍不完整,阻碍了基因组资源的全面研究和广泛应用。
- 研究材料及方法:研究随机选取 6 株成熟生菜植株,分为 2 个生物学重复组,每组 3 株。用超纯水清洗 3 次后,分别切成根、茎、叶 3 部分,进行 DNBSEQ 转录组测序以及多倍通量全长转录组测序(HIT-ISOseq)。
主要发现:
1) HIT-ISOseq 改进生菜的参考转录本注释和功能注释
利用 PacBio Sequel II SMRT Cell 8 M 测序芯片共获得 15.79 M reads,平均每个 CCS reads 有 3.46 个全长非嵌合序列(FLNC)。每个样本平均获得 2.55 M 到 2.74 M FLNC reads,平均读长范围从 736.31 bp到 779.97 bp。HIT-ISOseq 测序结果共鉴定出 31,297 个基因和 69,973 个转录本,每个样本的基因数量为 23,550 至 27,629 个,转录本数量为 54,711 至 58,998 个。
2) 定量一致性结果
从 HIT-ISOseq 测序数据得出的基因和转录本表达与 DNBSEQ (PE150) 测序获得的结果具有高度相关性,从而验证了基因表达定量的一致性。
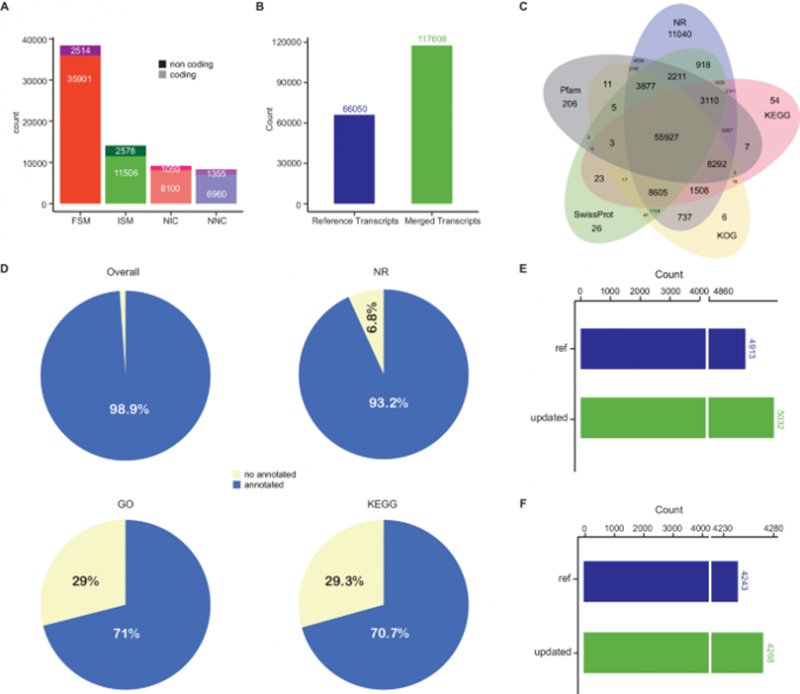
图1 HIT-ISOseq 更新生菜参考注释
3) HIT-ISOseq 揭示生菜中组织特异性表达基因及转录本
通过 HIT-ISOseq 测序数据鉴定到生菜中组织特异性表达的 2,611 个基因以及 4,842 个转录本。此外,研究者鉴定到 18 种表现出与相应基因表达模式不同的转录本,并对这些转录本进行了 qPCR 验证。
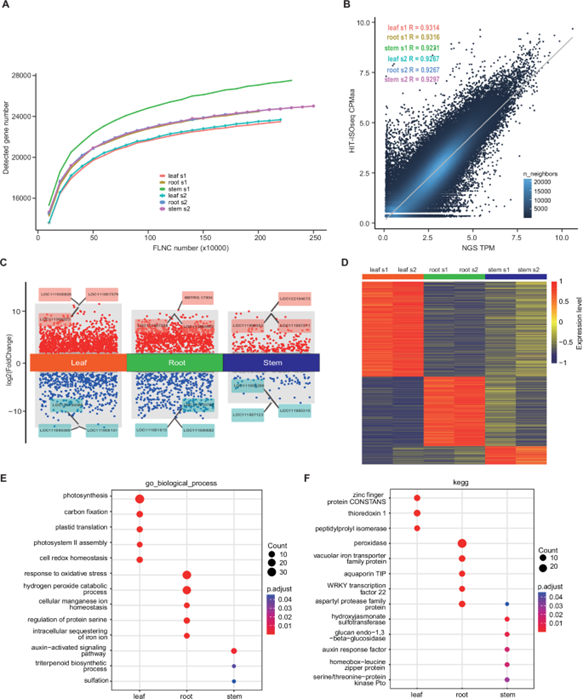
图2 HIT-ISOseq 揭示生菜中组织特异性表达基因
案例二:Poly(A)长度控制对精子发生过程中转录与翻译的调控作用[2]
发表杂志:Development(IF=6.862)
发表时间: 2022-6-15
研究方法:poly(A) 全长转录组测序、Ribo-seq
主要结论:
为探究精子细胞分化过程中 Poly(A) 长度动态变化与 mRNA 稳定性和翻译延迟的关系,作者通过构建 PAPA-seq 文库,使用 Poly(U) 聚合酶将 G、I 核苷酸加入到 mRNA 的 3′ 端,并进行反转录生成含有全长 Poly(A) 尾的 cDNA,使用 PacBio 平台测序检测精子发生过程中粗线期精母细胞(pachytene spermatocytes)、圆形(round)和长形精子细胞(elongating spermatids)以及精子的 Poly(A) 长度及 3’ UTR 长度变化(如图3);
PAPA-seq 结果显示,当粗线期精母细胞发育成圆形和长形精子细胞时,转录本全长呈缩短趋势,且 mRNAs 的 3’ UTRs 及 5’ UTRs 逐渐缩短。但 Poly(A) 长度则呈双相方式动态变化,从粗线期精母细胞到圆形精子细胞,Poly(A) 尾长度逐渐增加,这可能有助于延长 mRNA 半衰期,并在精子发生后期保护 mRNAs 以延迟翻译。相反,从圆形精子到长形精子过程中,Poly(A) 的长度逐渐变短,表明转录本 Poly(A) 长度变化与精子发生过程中的翻译延迟同步。
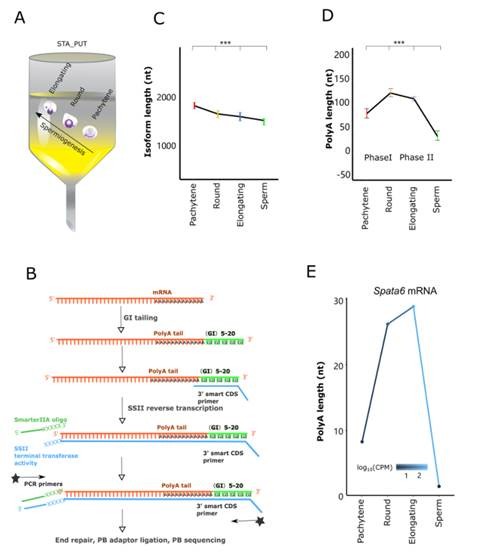
图3 Poly(A) 全长转录组深度测序显示精子发生过程中的 Poly(A) 长度动态变化
为了进一步探讨 Poly(A) 长度对 mRNA 翻译抑制和激活的影响,研究团队通过 PAPA-seq,发现定位在 RNP 的 mRNAs 的平均 Poly(A) 长度仅为定位在含多核糖体中(polysome)mRNA 的 1/40,这表明 mRNA 的去腺苷酸化与其在 RNPs 的定位之间存在强烈的关联。通过分析 RNP和 polysome 中的 miRNA-mRNA 靶向关系发现,在粗线期精母细胞中,与 polysome 转录本相比,RNP 富集型转录本中的 miRNA 靶点更集中在 3'端,精子发生进展到圆形和长形精子细胞阶段时,目标转录本 3' UTRs 上的 miRNA 靶点逐渐减少并最终消失,这与长形精子细胞中 RNP 的 mRNAs 释放相一致,表明 miRNAs 驱动 mRNAs 进入 RNPs 进行去腺苷酸化。具有较长的 Poly(A) 尾(>50 nt)的转录本从 RNP 组分转移到 polysome 组分,而具有较短的 Poly(A) 尾(<5 nt)的转录本往往被隔离在核糖核蛋白(RNP)颗粒中,以进行翻译抑制并保持稳定。
并且,通过 Drosha 条件敲除(cKO)小鼠模型及 miR-506 基因敲除(KO)小鼠系实验以消除 miRNA 的产生,验证 miRNA 通过去腺苷酸化缩短其靶 mRNA 的 Poly(A)长度,从而将它们隔离在 RNP 颗粒中以延迟翻译。
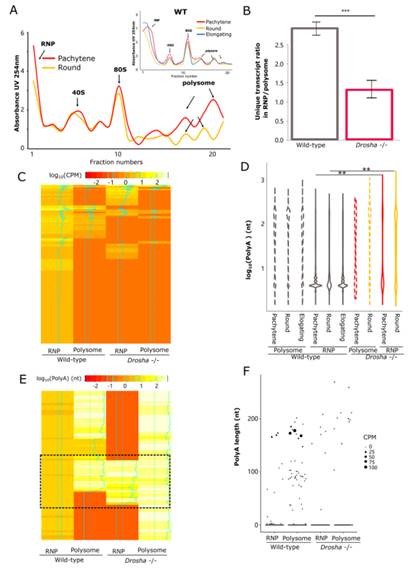
图4 miRNA 缺失对 RNP 颗粒和多核糖体组分中 Poly(A) 长度分布的影响
参考文献:
[1] Shi, ZX., Xiang, L., Zhao, HM. et al. High-throughput single-molecule long-read RNA sequencing analysis of tissue-specific genes and isoforms in lettuce (Lactuca sativa L.). Commun Biol 7, 920 (2024). https://doi.org/10.1038/s42003-024-06598-4.
[2] Guo, Mei et al. “Uncoupling transcription and translation through miRNA-dependent poly(A) length control in haploid male germ cells.” Development (Cambridge, England) vol. 149,12 (2022): dev199573. doi:10.1242/dev.199573.
多倍通量全长转录组
为了充分利用 PacBio 测序长读长优势,华大科技研发了多倍通量全长转录组专利技术,在建库过程中将多个转录本首尾相连构建多倍通量文库;通过 CCS 测序,一条 CCS read 可拆分得到多条转录本,大幅提升 RacBio 测序全长 reads 的获得率。
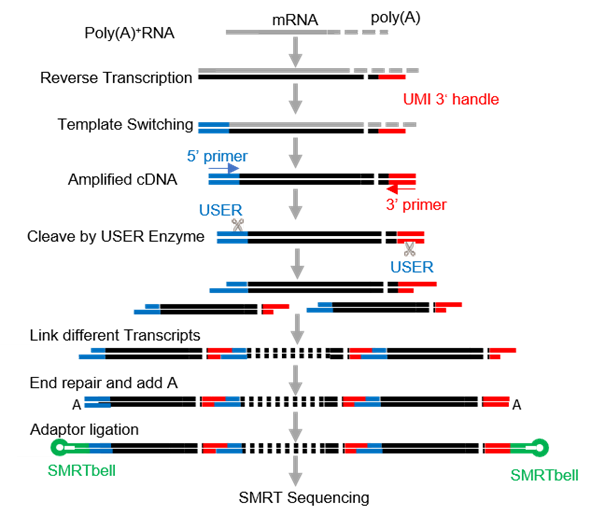
图1 多倍通量全长转录组建库示意图
技术优势
多倍通量全长转录组将单个 cell 的有效 reads 数提升至 3-6 倍以上;在成本不变的情况下,可得到更多的转录本检测数目;并结合 UMI 实现准确的基因/转录本定量。
1) 有效 reads 数提升 3-6 倍
同样的测序数据量,多倍通量全长转录组检测到的 reads 数是常规全长转录组的 3-6 倍。
2) 测序准确度高——基因结构分析
多倍通量全长转录组测序,使用 PacBio 测序平台 CCS 测序模式,测序准确性较高。同一序列 CCS 测序得到的 full-pass 数会减少,但 Isoform 校正过程中可用的 reads 数会增加,最终得到的序列准确度并不会降低,适用于转录本结构分析。
各物种的转录本多样且复杂,绝大多数真核生物基因都存在多种剪切形式,全长转录组无需打断直接读取从 5' 端到 3' 端的序列,可有效识别可变剪接(AS)、可选择性多聚腺苷酸化(APA)、基因家族、融合基因等,是基因结构分析的有效方式。
3) UMI 和多倍通量完美结合,基因定量结果更准确
UMI(Unique molecular identifier,特异性分子标识符)和多倍通量完美结合,基因定量结果和短读长平台一致性好,测序结果也可以做基因定量。
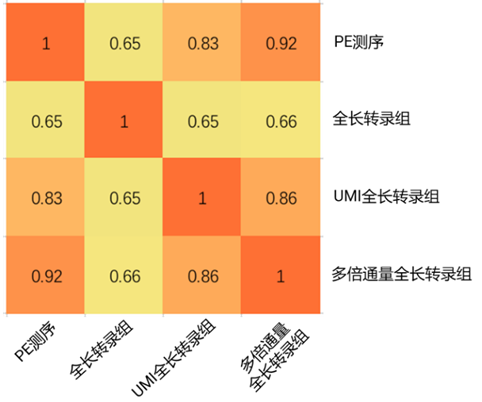
图2 基因定量一致性(spearman)
产品策略
建库:构建多倍通量全长转录组文库;
测序:利用 Revio 平台进行 CCS 测序,推荐测序 5M s-reads 数据量以上;
分析:根据有无参考基因组分为 Ref(有参)和 de novo(无参)分析。
表1. 测序数据量推荐
|
研究目标 |
稀有转录本异构体 发现、定量 |
中、高表达转录本异构体 发现、定量 |
鉴定高表达基因、 完善转录本注释 |
|
饱和度曲线数据表现 |
饱和度曲线整体趋于平台期 |
发现更多低/中/高表达基因和复杂转录本 |
基因及转录本检出率约80% |
|
推荐测序数据量 |
10M -20M |
5M |
~5M |
|
推荐样本类型 |
复杂转录本结构样本 |
肿瘤、疾病样本 |
动植物样本 |
Poly(A) 全长转录组
华大科技 poly(A)+ 全长转录组测序,基于 PacBio 长读长测序平台,可同时得到高准确度的全长转录本序列和 poly(A) 长度及碱基情况。测序数据除可用于分析可变剪接、融合基因、鉴定新异构体,精确定量转录本表达水平之外,更能全面检测 Poly(A) 长度和碱基类型。
助您深入研究 Poly(A) 在转录本表达中的调控作用,从 RNA 整体水平上研究 poly(A) 长度与转录本及非编码区的关系,实现从结果(基因表达量,结构变异)到原因(poly(A)调控机制)的全方位研究!
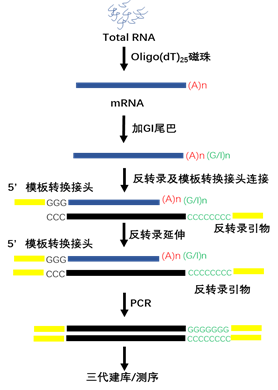
图3 poly(A) 全长转录组建库方法示意图
Poly(A)+ 全长转录组特色分析
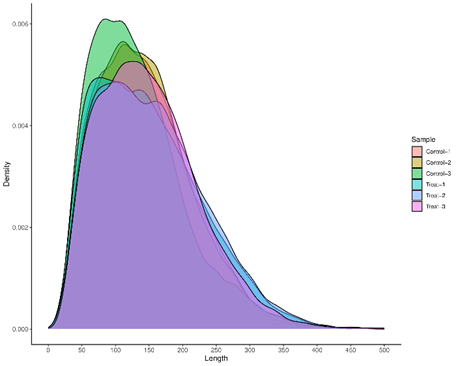
图4 所有样品的 poly(A) 长度分布图
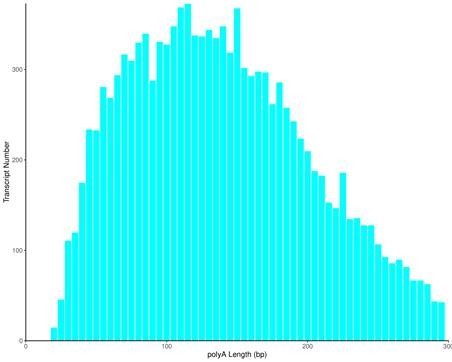
图5 各样本 poly(A) 长度分布图
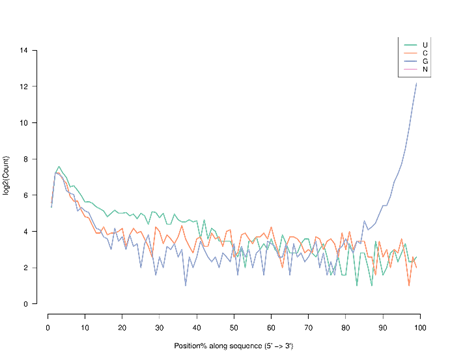
图6 poly(A) 非 A 碱基含量图
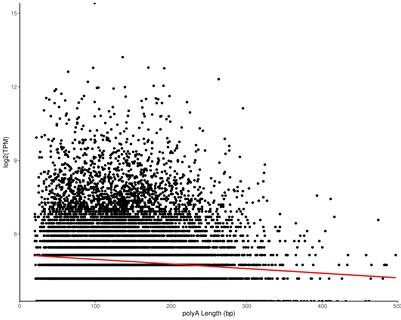
图7 poly(A) 长度与转录本表达量的关系图
RNA样本送样建议
PacBio全长转录组
|
样本类型 |
总量 |
浓度 |
RIN |
28S/18S |
基线和5S |
OD |
|
Total RNA |
1.5 µg |
285 ng/µL |
RIN≥7.5 |
28S/18S≥1.2 |
基线平整, 5S峰正常 |
OD260/280≥1.8 OD260/230≥1.6 |
组织样本送样建议
|
组织类型 |
送样量(提取RNA) |
注意事项 |
|
新鲜动物组织干重 |
≥100 mg |
|
|
新鲜植物组织干重 |
≥200 mg |
|
|
新鲜培养细胞 |
≥1×106 个 |
|
|
全血(哺乳动物) |
≥2.5 mL 淋巴细胞 ≥2.5 mL Paxgene Blood RNA
tube收集的全血 |
禁止使用肝素抗凝管采血 |
|
全血(非哺乳动物) |
≥0.2 mL RNAprotect® Animal
Blood Tubes收集的全血 |
禁止使用肝素抗凝管采血 |
|
菌体 |
≥2×107--2×108
个 |
若具有传染性需进行灭活处理 |
Q1:多倍通量全长转录组测序会不会影响转录本长度分布?
A1:不会。
多倍通量全长转录组技术将不同长度的转录本随机连接,无长度偏向性,可无差异检测到各个长度范围的转录本。如下图所示,和常规文库相比,多倍通量全长转录组文库的检测结果并无明显偏向性。
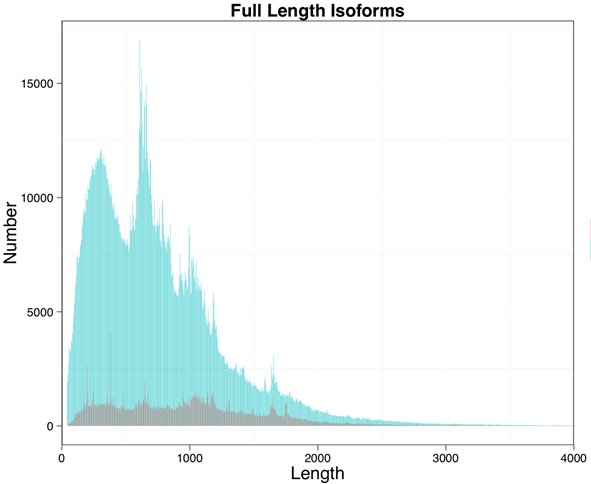
图1 全长转录本长度分布图
Q2:多倍通量全长转录组技术得到的reads准确度是否会降低?
A2: 不会。
测序长度越长,CCS 后单条序列准确度越高。PacBio 测序中构建哑铃型文库,在插入片段比酶读长短的情况下,通过环形比对测序(Circular Consensus Sequencing, CCS),同一个片段可以被循环测到很多次,这在很大程度增加最终得到的序列准确性。

图2 CCS 测序示意图。
分析过程中,通过 reads 聚类和 Isoform 校正进一步提升 Isoform 准确度。在 PacBio 数据分析中,为了提高最终得到的转录本序列的准确度,得到 CCS 序列之后会进行 reads 聚类和 Isoform 校正,进一步提高 Isoform 准确度,同一 Isoform 测到的条数越多,校正后的准确度越高。这一步可以通过阈值设定,筛选准确度达到要求的用于后续分析,目前一般将 Isoform 校正后准确度在 0.99 以上判定为高质量 Isoform。
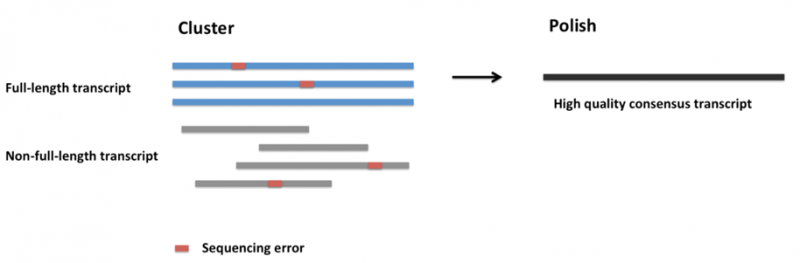
图3 Isoform校正过程示意图。
多倍通量全长转录组产品,同一序列 CCS 测序得到的 full-pass 数会减少,但 Isoform 校正过程中可用的 reads 数会相应增加,最终得到的序列准确度并不会降低。
Q3:多倍通量全长转录组产品能不能用来做基因/转录本定量?
A3:可以
多倍通量全长转录组产品结合了多倍通量和另外一项华大特有技术(UMI),在提高有效数据量的同时通过 UMI 对每一条转录本分子进行标记,可以排除建库过程的 PCR 偏向性,定量结果可达到和短读长平台基因定量相当的水平。
Q4:多倍通量全长转录组产品需要构建多个文库吗?
A4:不需要。
多倍通量全长转录组技术在构建哑铃型文库之前,将不同长度的转录本进行随机连接,并通过筛选得到有效连接产物,测序 loading 偏差小。
Q5:全长转录组目前能接原核转录组吗?
A5: 不能。目前全长转录组主要针对真核转录组,如果需要做原核转录组或 LncRNA,需个性化沟通。